Get trending papers in your email inbox once a day!
Get trending papers in your email inbox!
Subscribe





ShortGPT: Layers in Large Language Models are More Redundant Than You Expect
As Large Language Models (LLMs) continue to advance in performance, their size has escalated significantly, with current LLMs containing billions or even trillions of parameters. However, in this study, we discovered that many layers of LLMs exhibit high similarity, and some layers play a negligible role in network functionality. Based on this observation, we define a metric called Block Influence (BI) to gauge the significance of each layer in LLMs. We then propose a straightforward pruning approach: layer removal, in which we directly delete the redundant layers in LLMs based on their BI scores. Experiments demonstrate that our method, which we call ShortGPT, significantly outperforms previous state-of-the-art (SOTA) methods in model pruning. Moreover, ShortGPT is orthogonal to quantization-like methods, enabling further reduction in parameters and computation. The ability to achieve better results through simple layer removal, as opposed to more complex pruning techniques, suggests a high degree of redundancy in the model architecture.

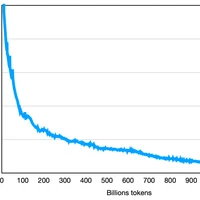
Scaling LLM Test-Time Compute Optimally can be More Effective than Scaling Model Parameters
Enabling LLMs to improve their outputs by using more test-time computation is a critical step towards building generally self-improving agents that can operate on open-ended natural language. In this paper, we study the scaling of inference-time computation in LLMs, with a focus on answering the question: if an LLM is allowed to use a fixed but non-trivial amount of inference-time compute, how much can it improve its performance on a challenging prompt? Answering this question has implications not only on the achievable performance of LLMs, but also on the future of LLM pretraining and how one should tradeoff inference-time and pre-training compute. Despite its importance, little research attempted to understand the scaling behaviors of various test-time inference methods. Moreover, current work largely provides negative results for a number of these strategies. In this work, we analyze two primary mechanisms to scale test-time computation: (1) searching against dense, process-based verifier reward models; and (2) updating the model's distribution over a response adaptively, given the prompt at test time. We find that in both cases, the effectiveness of different approaches to scaling test-time compute critically varies depending on the difficulty of the prompt. This observation motivates applying a "compute-optimal" scaling strategy, which acts to most effectively allocate test-time compute adaptively per prompt. Using this compute-optimal strategy, we can improve the efficiency of test-time compute scaling by more than 4x compared to a best-of-N baseline. Additionally, in a FLOPs-matched evaluation, we find that on problems where a smaller base model attains somewhat non-trivial success rates, test-time compute can be used to outperform a 14x larger model.


More Agents Is All You Need
We find that, simply via a sampling-and-voting method, the performance of large language models (LLMs) scales with the number of agents instantiated. Also, this method is orthogonal to existing complicated methods to further enhance LLMs, while the degree of enhancement is correlated to the task difficulty. We conduct comprehensive experiments on a wide range of LLM benchmarks to verify the presence of our finding, and to study the properties that can facilitate its occurrence. Our code is publicly available at: https://anonymous.4open.science/r/more_agent_is_all_you_need.
Multiple Choice Questions: Reasoning Makes Large Language Models (LLMs) More Self-Confident Even When They Are Wrong
One of the most widely used methods to evaluate LLMs are Multiple Choice Question (MCQ) tests. MCQ benchmarks enable the testing of LLM knowledge on almost any topic at scale as the results can be processed automatically. To help the LLM answer, a few examples called few shots can be included in the prompt. Moreover, the LLM can be asked to answer the question directly with the selected option or to first provide the reasoning and then the selected answer, which is known as chain of thought. In addition to checking whether the selected answer is correct, the evaluation can look at the LLM-estimated probability of its response as an indication of the confidence of the LLM in the response. In this paper, we study how the LLM confidence in its answer depends on whether the model has been asked to answer directly or to provide the reasoning before answering. The results of the evaluation of questions on a wide range of topics in seven different models show that LLMs are more confident in their answers when they provide reasoning before the answer. This occurs regardless of whether the selected answer is correct. Our hypothesis is that this behavior is due to the reasoning that modifies the probability of the selected answer, as the LLM predicts the answer based on the input question and the reasoning that supports the selection made. Therefore, LLM estimated probabilities seem to have intrinsic limitations that should be understood in order to use them in evaluation procedures. Interestingly, the same behavior has been observed in humans, for whom explaining an answer increases confidence in its correctness.




LIMA: Less Is More for Alignment
Large language models are trained in two stages: (1) unsupervised pretraining from raw text, to learn general-purpose representations, and (2) large scale instruction tuning and reinforcement learning, to better align to end tasks and user preferences. We measure the relative importance of these two stages by training LIMA, a 65B parameter LLaMa language model fine-tuned with the standard supervised loss on only 1,000 carefully curated prompts and responses, without any reinforcement learning or human preference modeling. LIMA demonstrates remarkably strong performance, learning to follow specific response formats from only a handful of examples in the training data, including complex queries that range from planning trip itineraries to speculating about alternate history. Moreover, the model tends to generalize well to unseen tasks that did not appear in the training data. In a controlled human study, responses from LIMA are either equivalent or strictly preferred to GPT-4 in 43% of cases; this statistic is as high as 58% when compared to Bard and 65% versus DaVinci003, which was trained with human feedback. Taken together, these results strongly suggest that almost all knowledge in large language models is learned during pretraining, and only limited instruction tuning data is necessary to teach models to produce high quality output.




More Documents, Same Length: Isolating the Challenge of Multiple Documents in RAG
Retrieval-augmented generation (RAG) provides LLMs with relevant documents. Although previous studies noted that retrieving many documents can degrade performance, they did not isolate how the quantity of documents affects performance while controlling for context length. We evaluate various language models on custom datasets derived from a multi-hop QA task. We keep the context length and position of relevant information constant while varying the number of documents, and find that increasing the document count in RAG settings poses significant challenges for LLMs. Additionally, our results indicate that processing multiple documents is a separate challenge from handling long contexts. We also make the datasets and code available: https://github.com/shaharl6000/MoreDocsSameLen .

Scaling Laws in Patchification: An Image Is Worth 50,176 Tokens And More
Since the introduction of Vision Transformer (ViT), patchification has long been regarded as a de facto image tokenization approach for plain visual architectures. By compressing the spatial size of images, this approach can effectively shorten the token sequence and reduce the computational cost of ViT-like plain architectures. In this work, we aim to thoroughly examine the information loss caused by this patchification-based compressive encoding paradigm and how it affects visual understanding. We conduct extensive patch size scaling experiments and excitedly observe an intriguing scaling law in patchification: the models can consistently benefit from decreased patch sizes and attain improved predictive performance, until it reaches the minimum patch size of 1x1, i.e., pixel tokenization. This conclusion is broadly applicable across different vision tasks, various input scales, and diverse architectures such as ViT and the recent Mamba models. Moreover, as a by-product, we discover that with smaller patches, task-specific decoder heads become less critical for dense prediction. In the experiments, we successfully scale up the visual sequence to an exceptional length of 50,176 tokens, achieving a competitive test accuracy of 84.6% with a base-sized model on the ImageNet-1k benchmark. We hope this study can provide insights and theoretical foundations for future works of building non-compressive vision models. Code is available at https://github.com/wangf3014/Patch_Scaling.



MoDec-GS: Global-to-Local Motion Decomposition and Temporal Interval Adjustment for Compact Dynamic 3D Gaussian Splatting
3D Gaussian Splatting (3DGS) has made significant strides in scene representation and neural rendering, with intense efforts focused on adapting it for dynamic scenes. Despite delivering remarkable rendering quality and speed, existing methods struggle with storage demands and representing complex real-world motions. To tackle these issues, we propose MoDecGS, a memory-efficient Gaussian splatting framework designed for reconstructing novel views in challenging scenarios with complex motions. We introduce GlobaltoLocal Motion Decomposition (GLMD) to effectively capture dynamic motions in a coarsetofine manner. This approach leverages Global Canonical Scaffolds (Global CS) and Local Canonical Scaffolds (Local CS), extending static Scaffold representation to dynamic video reconstruction. For Global CS, we propose Global Anchor Deformation (GAD) to efficiently represent global dynamics along complex motions, by directly deforming the implicit Scaffold attributes which are anchor position, offset, and local context features. Next, we finely adjust local motions via the Local Gaussian Deformation (LGD) of Local CS explicitly. Additionally, we introduce Temporal Interval Adjustment (TIA) to automatically control the temporal coverage of each Local CS during training, allowing MoDecGS to find optimal interval assignments based on the specified number of temporal segments. Extensive evaluations demonstrate that MoDecGS achieves an average 70% reduction in model size over stateoftheart methods for dynamic 3D Gaussians from realworld dynamic videos while maintaining or even improving rendering quality.



Towards 3D Molecule-Text Interpretation in Language Models
Language Models (LMs) have greatly influenced diverse domains. However, their inherent limitation in comprehending 3D molecular structures has considerably constrained their potential in the biomolecular domain. To bridge this gap, we focus on 3D molecule-text interpretation, and propose 3D-MoLM: 3D-Molecular Language Modeling. Specifically, 3D-MoLM enables an LM to interpret and analyze 3D molecules by equipping the LM with a 3D molecular encoder. This integration is achieved by a 3D molecule-text projector, bridging the 3D molecular encoder's representation space and the LM's input space. Moreover, to enhance 3D-MoLM's ability of cross-modal molecular understanding and instruction following, we meticulously curated a 3D molecule-centric instruction tuning dataset -- 3D-MoIT. Through 3D molecule-text alignment and 3D molecule-centric instruction tuning, 3D-MoLM establishes an integration of 3D molecular encoder and LM. It significantly surpasses existing baselines on downstream tasks, including molecule-text retrieval, molecule captioning, and more challenging open-text molecular QA tasks, especially focusing on 3D-dependent properties.




More Context, Less Distraction: Visual Classification by Inferring and Conditioning on Contextual Attributes
CLIP, as a foundational vision language model, is widely used in zero-shot image classification due to its ability to understand various visual concepts and natural language descriptions. However, how to fully leverage CLIP's unprecedented human-like understanding capabilities to achieve better zero-shot classification is still an open question. This paper draws inspiration from the human visual perception process: a modern neuroscience view suggests that in classifying an object, humans first infer its class-independent attributes (e.g., background and orientation) which help separate the foreground object from the background, and then make decisions based on this information. Inspired by this, we observe that providing CLIP with contextual attributes improves zero-shot classification and mitigates reliance on spurious features. We also observe that CLIP itself can reasonably infer the attributes from an image. With these observations, we propose a training-free, two-step zero-shot classification method named PerceptionCLIP. Given an image, it first infers contextual attributes (e.g., background) and then performs object classification conditioning on them. Our experiments show that PerceptionCLIP achieves better generalization, group robustness, and better interpretability. For example, PerceptionCLIP with ViT-L/14 improves the worst group accuracy by 16.5% on the Waterbirds dataset and by 3.5% on CelebA.




Less is More: Focus Attention for Efficient DETR
DETR-like models have significantly boosted the performance of detectors and even outperformed classical convolutional models. However, all tokens are treated equally without discrimination brings a redundant computational burden in the traditional encoder structure. The recent sparsification strategies exploit a subset of informative tokens to reduce attention complexity maintaining performance through the sparse encoder. But these methods tend to rely on unreliable model statistics. Moreover, simply reducing the token population hinders the detection performance to a large extent, limiting the application of these sparse models. We propose Focus-DETR, which focuses attention on more informative tokens for a better trade-off between computation efficiency and model accuracy. Specifically, we reconstruct the encoder with dual attention, which includes a token scoring mechanism that considers both localization and category semantic information of the objects from multi-scale feature maps. We efficiently abandon the background queries and enhance the semantic interaction of the fine-grained object queries based on the scores. Compared with the state-of-the-art sparse DETR-like detectors under the same setting, our Focus-DETR gets comparable complexity while achieving 50.4AP (+2.2) on COCO. The code is available at https://github.com/huawei-noah/noah-research/tree/master/Focus-DETR and https://gitee.com/mindspore/models/tree/master/research/cv/Focus-DETR.
Advancing Molecular Machine (Learned) Representations with Stereoelectronics-Infused Molecular Graphs
Molecular representation is a foundational element in our understanding of the physical world. Its importance ranges from the fundamentals of chemical reactions to the design of new therapies and materials. Previous molecular machine learning models have employed strings, fingerprints, global features, and simple molecular graphs that are inherently information-sparse representations. However, as the complexity of prediction tasks increases, the molecular representation needs to encode higher fidelity information. This work introduces a novel approach to infusing quantum-chemical-rich information into molecular graphs via stereoelectronic effects. We show that the explicit addition of stereoelectronic interactions significantly improves the performance of molecular machine learning models. Furthermore, stereoelectronics-infused representations can be learned and deployed with a tailored double graph neural network workflow, enabling its application to any downstream molecular machine learning task. Finally, we show that the learned representations allow for facile stereoelectronic evaluation of previously intractable systems, such as entire proteins, opening new avenues of molecular design.



MolReFlect: Towards In-Context Fine-grained Alignments between Molecules and Texts
Molecule discovery is a pivotal research field, impacting everything from the medicines we take to the materials we use. Recently, Large Language Models (LLMs) have been widely adopted in molecule understanding and generation, yet the alignments between molecules and their corresponding captions remain a significant challenge. Previous endeavours often treat the molecule as a general SMILES string or molecular graph, neglecting the fine-grained alignments between the molecular sub-structures and the descriptive textual phrases, which are crucial for accurate and explainable predictions. In this case, we introduce MolReFlect, a novel teacher-student framework designed to contextually perform the molecule-caption alignments in a fine-grained way. Our approach initially leverages a larger teacher LLM to label the detailed alignments by directly extracting critical phrases from molecule captions or SMILES strings and implying them to corresponding sub-structures or characteristics. To refine these alignments, we propose In-Context Selective Reflection, which retrieves previous extraction results as context examples for teacher LLM to reflect and lets a smaller student LLM select from in-context reflection and previous extraction results. Finally, we enhance the learning process of the student LLM through Chain-of-Thought In-Context Molecule Tuning, integrating the fine-grained alignments and the reasoning processes within the Chain-of-Thought format. Our experimental results demonstrate that MolReFlect enables LLMs like Mistral-7B to significantly outperform the previous baselines, achieving SOTA performance on the ChEBI-20 dataset. This advancement not only enhances the generative capabilities of LLMs in the molecule-caption translation task, but also contributes to a more explainable framework.


In-context Vectors: Making In Context Learning More Effective and Controllable Through Latent Space Steering
Large language models (LLMs) demonstrate emergent in-context learning capabilities, where they adapt to new tasks based on example demonstrations. However, in-context learning has seen limited effectiveness in many settings, is difficult to quantitatively control and takes up context window space. To overcome these limitations, we propose an alternative approach that recasts in-context learning as in-context vectors (ICV). Using ICV has two steps. We first use a forward pass on demonstration examples to create the in-context vector from the latent embedding of the LLM. This vector captures essential information about the intended task. On a new query, instead of adding demonstrations to the prompt, we shift the latent states of the LLM using the ICV. The ICV approach has several benefits: 1) it enables the LLM to more effectively follow the demonstration examples; 2) it's easy to control by adjusting the magnitude of the ICV; 3) it reduces the length of the prompt by removing the in-context demonstrations; 4) ICV is computationally much more efficient than fine-tuning. We demonstrate that ICV achieves better performance compared to standard in-context learning and fine-tuning on diverse tasks including safety, style transfer, role-playing and formatting. Moreover, we show that we can flexibly teach LLM to simultaneously follow different types of instructions by simple vector arithmetics on the corresponding ICVs.
ScaleLong: Towards More Stable Training of Diffusion Model via Scaling Network Long Skip Connection
In diffusion models, UNet is the most popular network backbone, since its long skip connects (LSCs) to connect distant network blocks can aggregate long-distant information and alleviate vanishing gradient. Unfortunately, UNet often suffers from unstable training in diffusion models which can be alleviated by scaling its LSC coefficients smaller. However, theoretical understandings of the instability of UNet in diffusion models and also the performance improvement of LSC scaling remain absent yet. To solve this issue, we theoretically show that the coefficients of LSCs in UNet have big effects on the stableness of the forward and backward propagation and robustness of UNet. Specifically, the hidden feature and gradient of UNet at any layer can oscillate and their oscillation ranges are actually large which explains the instability of UNet training. Moreover, UNet is also provably sensitive to perturbed input, and predicts an output distant from the desired output, yielding oscillatory loss and thus oscillatory gradient. Besides, we also observe the theoretical benefits of the LSC coefficient scaling of UNet in the stableness of hidden features and gradient and also robustness. Finally, inspired by our theory, we propose an effective coefficient scaling framework ScaleLong that scales the coefficients of LSC in UNet and better improves the training stability of UNet. Experimental results on four famous datasets show that our methods are superior to stabilize training and yield about 1.5x training acceleration on different diffusion models with UNet or UViT backbones. Code: https://github.com/sail-sg/ScaleLong
Molecular Language Model as Multi-task Generator
Molecule generation with desired properties has grown immensely in popularity by disruptively changing the way scientists design molecular structures and providing support for chemical and materials design. However, despite the promising outcome, previous machine learning-based deep generative models suffer from a reliance on complex, task-specific fine-tuning, limited dimensional latent spaces, or the quality of expert rules. In this work, we propose MolGen, a pre-trained molecular language model that effectively learns and shares knowledge across multiple generation tasks and domains. Specifically, we pre-train MolGen with the chemical language SELFIES on more than 100 million unlabelled molecules. We further propose multi-task molecular prefix tuning across several molecular generation tasks and different molecular domains (synthetic & natural products) with a self-feedback mechanism. Extensive experiments show that MolGen can obtain superior performances on well-known molecular generation benchmark datasets. The further analysis illustrates that MolGen can accurately capture the distribution of molecules, implicitly learn their structural characteristics, and efficiently explore the chemical space with the guidance of multi-task molecular prefix tuning. Codes, datasets, and the pre-trained model will be available in https://github.com/zjunlp/MolGen.



More Expressive Attention with Negative Weights
We propose a novel attention mechanism, named Cog Attention, that enables attention weights to be negative for enhanced expressiveness, which stems from two key factors: (1) Cog Attention can shift the token deletion and copying function from a static OV matrix to dynamic QK inner products, with the OV matrix now focusing more on refinement or modification. The attention head can simultaneously delete, copy, or retain tokens by assigning them negative, positive, or minimal attention weights, respectively. As a result, a single attention head becomes more flexible and expressive. (2) Cog Attention improves the model's robustness against representational collapse, which can occur when earlier tokens are over-squashed into later positions, leading to homogeneous representations. Negative weights reduce effective information paths from earlier to later tokens, helping to mitigate this issue. We develop Transformer-like models which use Cog Attention as attention modules, including decoder-only models for language modeling and U-ViT diffusion models for image generation. Experiments show that models using Cog Attention exhibit superior performance compared to those employing traditional softmax attention modules. Our approach suggests a promising research direction for rethinking and breaking the entrenched constraints of traditional softmax attention, such as the requirement for non-negative weights.


ReactXT: Understanding Molecular "Reaction-ship" via Reaction-Contextualized Molecule-Text Pretraining
Molecule-text modeling, which aims to facilitate molecule-relevant tasks with a textual interface and textual knowledge, is an emerging research direction. Beyond single molecules, studying reaction-text modeling holds promise for helping the synthesis of new materials and drugs. However, previous works mostly neglect reaction-text modeling: they primarily focus on modeling individual molecule-text pairs or learning chemical reactions without texts in context. Additionally, one key task of reaction-text modeling -- experimental procedure prediction -- is less explored due to the absence of an open-source dataset. The task is to predict step-by-step actions of conducting chemical experiments and is crucial to automating chemical synthesis. To resolve the challenges above, we propose a new pretraining method, ReactXT, for reaction-text modeling, and a new dataset, OpenExp, for experimental procedure prediction. Specifically, ReactXT features three types of input contexts to incrementally pretrain LMs. Each of the three input contexts corresponds to a pretraining task to improve the text-based understanding of either reactions or single molecules. ReactXT demonstrates consistent improvements in experimental procedure prediction and molecule captioning and offers competitive results in retrosynthesis. Our code is available at https://github.com/syr-cn/ReactXT.

Less is More: Fewer Interpretable Region via Submodular Subset Selection
Image attribution algorithms aim to identify important regions that are highly relevant to model decisions. Although existing attribution solutions can effectively assign importance to target elements, they still face the following challenges: 1) existing attribution methods generate inaccurate small regions thus misleading the direction of correct attribution, and 2) the model cannot produce good attribution results for samples with wrong predictions. To address the above challenges, this paper re-models the above image attribution problem as a submodular subset selection problem, aiming to enhance model interpretability using fewer regions. To address the lack of attention to local regions, we construct a novel submodular function to discover more accurate small interpretation regions. To enhance the attribution effect for all samples, we also impose four different constraints on the selection of sub-regions, i.e., confidence, effectiveness, consistency, and collaboration scores, to assess the importance of various subsets. Moreover, our theoretical analysis substantiates that the proposed function is in fact submodular. Extensive experiments show that the proposed method outperforms SOTA methods on two face datasets (Celeb-A and VGG-Face2) and one fine-grained dataset (CUB-200-2011). For correctly predicted samples, the proposed method improves the Deletion and Insertion scores with an average of 4.9% and 2.5% gain relative to HSIC-Attribution. For incorrectly predicted samples, our method achieves gains of 81.0% and 18.4% compared to the HSIC-Attribution algorithm in the average highest confidence and Insertion score respectively. The code is released at https://github.com/RuoyuChen10/SMDL-Attribution.
Generative Modeling of Molecular Dynamics Trajectories
Molecular dynamics (MD) is a powerful technique for studying microscopic phenomena, but its computational cost has driven significant interest in the development of deep learning-based surrogate models. We introduce generative modeling of molecular trajectories as a paradigm for learning flexible multi-task surrogate models of MD from data. By conditioning on appropriately chosen frames of the trajectory, we show such generative models can be adapted to diverse tasks such as forward simulation, transition path sampling, and trajectory upsampling. By alternatively conditioning on part of the molecular system and inpainting the rest, we also demonstrate the first steps towards dynamics-conditioned molecular design. We validate the full set of these capabilities on tetrapeptide simulations and show that our model can produce reasonable ensembles of protein monomers. Altogether, our work illustrates how generative modeling can unlock value from MD data towards diverse downstream tasks that are not straightforward to address with existing methods or even MD itself. Code is available at https://github.com/bjing2016/mdgen.
More Consideration for the Perceptron
In this paper, we introduce the gated perceptron, an enhancement of the conventional perceptron, which incorporates an additional input computed as the product of the existing inputs. This allows the perceptron to capture non-linear interactions between features, significantly improving its ability to classify and regress on complex datasets. We explore its application in both linear and non-linear regression tasks using the Iris dataset, as well as binary and multi-class classification problems, including the PIMA Indian dataset and Breast Cancer Wisconsin dataset. Our results demonstrate that the gated perceptron can generate more distinct decision regions compared to traditional perceptrons, enhancing its classification capabilities, particularly in handling non-linear data. Performance comparisons show that the gated perceptron competes with state-of-the-art classifiers while maintaining a simple architecture.

MoreHopQA: More Than Multi-hop Reasoning
Most existing multi-hop datasets are extractive answer datasets, where the answers to the questions can be extracted directly from the provided context. This often leads models to use heuristics or shortcuts instead of performing true multi-hop reasoning. In this paper, we propose a new multi-hop dataset, MoreHopQA, which shifts from extractive to generative answers. Our dataset is created by utilizing three existing multi-hop datasets: HotpotQA, 2WikiMultihopQA, and MuSiQue. Instead of relying solely on factual reasoning, we enhance the existing multi-hop questions by adding another layer of questioning that involves one, two, or all three of the following types of reasoning: commonsense, arithmetic, and symbolic. Our dataset is created through a semi-automated process, resulting in a dataset with 1,118 samples that have undergone human verification. We then use our dataset to evaluate five different large language models: Mistral 7B, Gemma 7B, Llama 3 (8B and 70B), and GPT-4. We also design various cases to analyze the reasoning steps in the question-answering process. Our results show that models perform well on initial multi-hop questions but struggle with our extended questions, indicating that our dataset is more challenging than previous ones. Our analysis of question decomposition reveals that although models can correctly answer questions, only a portion - 38.7% for GPT-4 and 33.4% for Llama3-70B - achieve perfect reasoning, where all corresponding sub-questions are answered correctly. Evaluation code and data are available at https://github.com/Alab-NII/morehopqa
UEMM-Air: Make Unmanned Aerial Vehicles Perform More Multi-modal Tasks
The development of multi-modal learning for Unmanned Aerial Vehicles (UAVs) typically relies on a large amount of pixel-aligned multi-modal image data. However, existing datasets face challenges such as limited modalities, high construction costs, and imprecise annotations. To this end, we propose a synthetic multi-modal UAV-based multi-task dataset, UEMM-Air. Specifically, we simulate various UAV flight scenarios and object types using the Unreal Engine (UE). Then we design the UAV's flight logic to automatically collect data from different scenarios, perspectives, and altitudes. Furthermore, we propose a novel heuristic automatic annotation algorithm to generate accurate object detection labels. Finally, we utilize labels to generate text descriptions of images to make our UEMM-Air support more cross-modality tasks. In total, our UEMM-Air consists of 120k pairs of images with 6 modalities and precise annotations. Moreover, we conduct numerous experiments and establish new benchmark results on our dataset. We also found that models pre-trained on UEMM-Air exhibit better performance on downstream tasks compared to other similar datasets. The dataset is publicly available (https://github.com/1e12Leon/UEMM-Air) to support the research of multi-modal tasks on UAVs.
More Compute Is What You Need
Large language model pre-training has become increasingly expensive, with most practitioners relying on scaling laws to allocate compute budgets for model size and training tokens, commonly referred to as Compute-Optimal or Chinchilla Optimal. In this paper, we hypothesize a new scaling law that suggests model performance depends mostly on the amount of compute spent for transformer-based models, independent of the specific allocation to model size and dataset size. Using this unified scaling law, we predict that (a) for inference efficiency, training should prioritize smaller model sizes and larger training datasets, and (b) assuming the exhaustion of available web datasets, scaling the model size might be the only way to further improve model performance.

MORE-3S:Multimodal-based Offline Reinforcement Learning with Shared Semantic Spaces
Drawing upon the intuition that aligning different modalities to the same semantic embedding space would allow models to understand states and actions more easily, we propose a new perspective to the offline reinforcement learning (RL) challenge. More concretely, we transform it into a supervised learning task by integrating multimodal and pre-trained language models. Our approach incorporates state information derived from images and action-related data obtained from text, thereby bolstering RL training performance and promoting long-term strategic thinking. We emphasize the contextual understanding of language and demonstrate how decision-making in RL can benefit from aligning states' and actions' representation with languages' representation. Our method significantly outperforms current baselines as evidenced by evaluations conducted on Atari and OpenAI Gym environments. This contributes to advancing offline RL performance and efficiency while providing a novel perspective on offline RL.Our code and data are available at https://github.com/Zheng0428/MORE_.



Quantum-Inspired Machine Learning for Molecular Docking
Molecular docking is an important tool for structure-based drug design, accelerating the efficiency of drug development. Complex and dynamic binding processes between proteins and small molecules require searching and sampling over a wide spatial range. Traditional docking by searching for possible binding sites and conformations is computationally complex and results poorly under blind docking. Quantum-inspired algorithms combining quantum properties and annealing show great advantages in solving combinatorial optimization problems. Inspired by this, we achieve an improved in blind docking by using quantum-inspired combined with gradients learned by deep learning in the encoded molecular space. Numerical simulation shows that our method outperforms traditional docking algorithms and deep learning-based algorithms over 10\%. Compared to the current state-of-the-art deep learning-based docking algorithm DiffDock, the success rate of Top-1 (RMSD<2) achieves an improvement from 33\% to 35\% in our same setup. In particular, a 6\% improvement is realized in the high-precision region(RMSD<1) on molecules data unseen in DiffDock, which demonstrates the well-generalized of our method.
Equivariant Scalar Fields for Molecular Docking with Fast Fourier Transforms
Molecular docking is critical to structure-based virtual screening, yet the throughput of such workflows is limited by the expensive optimization of scoring functions involved in most docking algorithms. We explore how machine learning can accelerate this process by learning a scoring function with a functional form that allows for more rapid optimization. Specifically, we define the scoring function to be the cross-correlation of multi-channel ligand and protein scalar fields parameterized by equivariant graph neural networks, enabling rapid optimization over rigid-body degrees of freedom with fast Fourier transforms. The runtime of our approach can be amortized at several levels of abstraction, and is particularly favorable for virtual screening settings with a common binding pocket. We benchmark our scoring functions on two simplified docking-related tasks: decoy pose scoring and rigid conformer docking. Our method attains similar but faster performance on crystal structures compared to the widely-used Vina and Gnina scoring functions, and is more robust on computationally predicted structures. Code is available at https://github.com/bjing2016/scalar-fields.

Multi-scale Iterative Refinement towards Robust and Versatile Molecular Docking
Molecular docking is a key computational tool utilized to predict the binding conformations of small molecules to protein targets, which is fundamental in the design of novel drugs. Despite recent advancements in geometric deep learning-based approaches leading to improvements in blind docking efficiency, these methods have encountered notable challenges, such as limited generalization performance on unseen proteins, the inability to concurrently address the settings of blind docking and site-specific docking, and the frequent occurrence of physical implausibilities such as inter-molecular steric clash. In this study, we introduce DeltaDock, a robust and versatile framework designed for efficient molecular docking to overcome these challenges. DeltaDock operates in a two-step process: rapid initial complex structures sampling followed by multi-scale iterative refinement of the initial structures. In the initial stage, to sample accurate structures with high efficiency, we develop a ligand-dependent binding site prediction model founded on large protein models and graph neural networks. This model is then paired with GPU-accelerated sampling algorithms. The sampled structures are updated using a multi-scale iterative refinement module that captures both protein-ligand atom-atom interactions and residue-atom interactions in the following stage. Distinct from previous geometric deep learning methods that are conditioned on the blind docking setting, DeltaDock demonstrates superior performance in both blind docking and site-specific docking settings. Comprehensive experimental results reveal that DeltaDock consistently surpasses baseline methods in terms of docking accuracy. Furthermore, it displays remarkable generalization capabilities and proficiency for predicting physically valid structures, thereby attesting to its robustness and reliability in various scenarios.
Can pruning make Large Language Models more efficient?
Transformer models have revolutionized natural language processing with their unparalleled ability to grasp complex contextual relationships. However, the vast number of parameters in these models has raised concerns regarding computational efficiency, environmental impact, and deployability on resource-limited platforms. To address these challenges, this paper investigates the application of weight pruning-a strategic reduction of model parameters based on their significance-as an optimization strategy for Transformer architectures. Through extensive experimentation, we explore various pruning methodologies, highlighting their impact on model performance, size, and computational demands. Our findings suggest that with judicious selection of pruning hyperparameters, significant reductions in model size are attainable without considerable compromise on performance. Moreover, when coupled with post-pruning fine-tuning strategies, some pruned models even exhibit enhanced generalization capabilities. This work seeks to bridge the gap between model efficiency and performance, paving the way for more scalable and environmentally responsible deep learning applications.

MDCS: More Diverse Experts with Consistency Self-distillation for Long-tailed Recognition
Recently, multi-expert methods have led to significant improvements in long-tail recognition (LTR). We summarize two aspects that need further enhancement to contribute to LTR boosting: (1) More diverse experts; (2) Lower model variance. However, the previous methods didn't handle them well. To this end, we propose More Diverse experts with Consistency Self-distillation (MDCS) to bridge the gap left by earlier methods. Our MDCS approach consists of two core components: Diversity Loss (DL) and Consistency Self-distillation (CS). In detail, DL promotes diversity among experts by controlling their focus on different categories. To reduce the model variance, we employ KL divergence to distill the richer knowledge of weakly augmented instances for the experts' self-distillation. In particular, we design Confident Instance Sampling (CIS) to select the correctly classified instances for CS to avoid biased/noisy knowledge. In the analysis and ablation study, we demonstrate that our method compared with previous work can effectively increase the diversity of experts, significantly reduce the variance of the model, and improve recognition accuracy. Moreover, the roles of our DL and CS are mutually reinforcing and coupled: the diversity of experts benefits from the CS, and the CS cannot achieve remarkable results without the DL. Experiments show our MDCS outperforms the state-of-the-art by 1% sim 2% on five popular long-tailed benchmarks, including CIFAR10-LT, CIFAR100-LT, ImageNet-LT, Places-LT, and iNaturalist 2018. The code is available at https://github.com/fistyee/MDCS.

More efficient manual review of automatically transcribed tabular data
Machine learning methods have proven useful in transcribing historical data. However, results from even highly accurate methods require manual verification and correction. Such manual review can be time-consuming and expensive, therefore the objective of this paper was to make it more efficient. Previously, we used machine learning to transcribe 2.3 million handwritten occupation codes from the Norwegian 1950 census with high accuracy (97%). We manually reviewed the 90,000 (3%) codes with the lowest model confidence. We allocated those 90,000 codes to human reviewers, who used our annotation tool to review the codes. To assess reviewer agreement, some codes were assigned to multiple reviewers. We then analyzed the review results to understand the relationship between accuracy improvements and effort. Additionally, we interviewed the reviewers to improve the workflow. The reviewers corrected 62.8% of the labels and agreed with the model label in 31.9% of cases. About 0.2% of the images could not be assigned a label, while for 5.1% the reviewers were uncertain, or they assigned an invalid label. 9,000 images were independently reviewed by multiple reviewers, resulting in an agreement of 86.43% and disagreement of 8.96%. We learned that our automatic transcription is biased towards the most frequent codes, with a higher degree of misclassification for the lowest frequency codes. Our interview findings show that the reviewers did internal quality control and found our custom tool well-suited. So, only one reviewer is needed, but they should report uncertainty.
The EarlyBIRD Catches the Bug: On Exploiting Early Layers of Encoder Models for More Efficient Code Classification
The use of modern Natural Language Processing (NLP) techniques has shown to be beneficial for software engineering tasks, such as vulnerability detection and type inference. However, training deep NLP models requires significant computational resources. This paper explores techniques that aim at achieving the best usage of resources and available information in these models. We propose a generic approach, EarlyBIRD, to build composite representations of code from the early layers of a pre-trained transformer model. We empirically investigate the viability of this approach on the CodeBERT model by comparing the performance of 12 strategies for creating composite representations with the standard practice of only using the last encoder layer. Our evaluation on four datasets shows that several early layer combinations yield better performance on defect detection, and some combinations improve multi-class classification. More specifically, we obtain a +2 average improvement of detection accuracy on Devign with only 3 out of 12 layers of CodeBERT and a 3.3x speed-up of fine-tuning. These findings show that early layers can be used to obtain better results using the same resources, as well as to reduce resource usage during fine-tuning and inference.



DiffDock: Diffusion Steps, Twists, and Turns for Molecular Docking
Predicting the binding structure of a small molecule ligand to a protein -- a task known as molecular docking -- is critical to drug design. Recent deep learning methods that treat docking as a regression problem have decreased runtime compared to traditional search-based methods but have yet to offer substantial improvements in accuracy. We instead frame molecular docking as a generative modeling problem and develop DiffDock, a diffusion generative model over the non-Euclidean manifold of ligand poses. To do so, we map this manifold to the product space of the degrees of freedom (translational, rotational, and torsional) involved in docking and develop an efficient diffusion process on this space. Empirically, DiffDock obtains a 38% top-1 success rate (RMSD<2A) on PDBBind, significantly outperforming the previous state-of-the-art of traditional docking (23%) and deep learning (20%) methods. Moreover, while previous methods are not able to dock on computationally folded structures (maximum accuracy 10.4%), DiffDock maintains significantly higher precision (21.7%). Finally, DiffDock has fast inference times and provides confidence estimates with high selective accuracy.

MoEC: Mixture of Expert Clusters
Sparsely Mixture of Experts (MoE) has received great interest due to its promising scaling capability with affordable computational overhead. MoE converts dense layers into sparse experts, and utilizes a gated routing network to make experts conditionally activated. However, as the number of experts grows, MoE with outrageous parameters suffers from overfitting and sparse data allocation. Such problems are especially severe on tasks with limited data, thus hindering the progress for MoE models to improve performance by scaling up. In this work, we propose Mixture of Expert Clusters - a general approach to enable expert layers to learn more diverse and appropriate knowledge by imposing variance-based constraints on the routing stage. We further propose a cluster-level expert dropout strategy specifically designed for the expert cluster structure. Our experiments reveal that MoEC could improve performance on machine translation and natural language understanding tasks, and raise the performance upper bound for scaling up experts under limited data. We also verify that MoEC plays a positive role in mitigating overfitting and sparse data allocation.
Less is More: Pay Less Attention in Vision Transformers
Transformers have become one of the dominant architectures in deep learning, particularly as a powerful alternative to convolutional neural networks (CNNs) in computer vision. However, Transformer training and inference in previous works can be prohibitively expensive due to the quadratic complexity of self-attention over a long sequence of representations, especially for high-resolution dense prediction tasks. To this end, we present a novel Less attention vIsion Transformer (LIT), building upon the fact that the early self-attention layers in Transformers still focus on local patterns and bring minor benefits in recent hierarchical vision Transformers. Specifically, we propose a hierarchical Transformer where we use pure multi-layer perceptrons (MLPs) to encode rich local patterns in the early stages while applying self-attention modules to capture longer dependencies in deeper layers. Moreover, we further propose a learned deformable token merging module to adaptively fuse informative patches in a non-uniform manner. The proposed LIT achieves promising performance on image recognition tasks, including image classification, object detection and instance segmentation, serving as a strong backbone for many vision tasks. Code is available at: https://github.com/zhuang-group/LIT



Molecular Graph Convolutions: Moving Beyond Fingerprints
Molecular "fingerprints" encoding structural information are the workhorse of cheminformatics and machine learning in drug discovery applications. However, fingerprint representations necessarily emphasize particular aspects of the molecular structure while ignoring others, rather than allowing the model to make data-driven decisions. We describe molecular "graph convolutions", a machine learning architecture for learning from undirected graphs, specifically small molecules. Graph convolutions use a simple encoding of the molecular graph---atoms, bonds, distances, etc.---which allows the model to take greater advantage of information in the graph structure. Although graph convolutions do not outperform all fingerprint-based methods, they (along with other graph-based methods) represent a new paradigm in ligand-based virtual screening with exciting opportunities for future improvement.
À la recherche du sens perdu: your favourite LLM might have more to say than you can understand
We report a peculiar observation that LLMs can assign hidden meanings to sequences that seem visually incomprehensible to humans: for example, a nonsensical phrase consisting of Byzantine musical symbols is recognized by gpt-4o as "say abracadabra". Moreover, some models can communicate using these sequences. Some of these meanings are hypothesized to partly originate in the massive spurious correlations due to BPE tokenization. We systematically evaluate the presence of such abilities in a wide range of models: Claude-3.5 Haiku, Claude-3.5 Sonnet (New and Old), Claude-3.7 Sonnet, gpt-4o mini, gpt-4o, o1-mini, Llama-3.3 70B, DeepSeek-R1-Distill-Lllama 70B, Qwen2.5 1.5B, Qwen2.5 32B, Phi-3.5 mini, GigaChat-Max, Vikhr-Llama-3.2 1B. We argue that this observation might have far-reaching consequences for both safety and security of the modern and future LLMs and systems that employ them. As an illustration, we show that applying this method in combination with simple templates is sufficient to jailbreak previous generation models, with ASR = 0.4 on gpt-4o mini. Our code and data artifacts are available at https://github.com/L3G5/llm-hidden-meanings
Molecular-driven Foundation Model for Oncologic Pathology
Foundation models are reshaping computational pathology by enabling transfer learning, where models pre-trained on vast datasets can be adapted for downstream diagnostic, prognostic, and therapeutic response tasks. Despite these advances, foundation models are still limited in their ability to encode the entire gigapixel whole-slide images without additional training and often lack complementary multimodal data. Here, we introduce Threads, a slide-level foundation model capable of generating universal representations of whole-slide images of any size. Threads was pre-trained using a multimodal learning approach on a diverse cohort of 47,171 hematoxylin and eosin (H&E)-stained tissue sections, paired with corresponding genomic and transcriptomic profiles - the largest such paired dataset to be used for foundation model development to date. This unique training paradigm enables Threads to capture the tissue's underlying molecular composition, yielding powerful representations applicable to a wide array of downstream tasks. In extensive benchmarking across 54 oncology tasks, including clinical subtyping, grading, mutation prediction, immunohistochemistry status determination, treatment response prediction, and survival prediction, Threads outperformed all baselines while demonstrating remarkable generalizability and label efficiency. It is particularly well suited for predicting rare events, further emphasizing its clinical utility. We intend to make the model publicly available for the broader community.
More Tokens, Lower Precision: Towards the Optimal Token-Precision Trade-off in KV Cache Compression
As large language models (LLMs) process increasing context windows, the memory usage of KV cache has become a critical bottleneck during inference. The mainstream KV compression methods, including KV pruning and KV quantization, primarily focus on either token or precision dimension and seldom explore the efficiency of their combination. In this paper, we comprehensively investigate the token-precision trade-off in KV cache compression. Experiments demonstrate that storing more tokens in the KV cache with lower precision, i.e., quantized pruning, can significantly enhance the long-context performance of LLMs. Furthermore, in-depth analysis regarding token-precision trade-off from a series of key aspects exhibit that, quantized pruning achieves substantial improvements in retrieval-related tasks and consistently performs well across varying input lengths. Moreover, quantized pruning demonstrates notable stability across different KV pruning methods, quantization strategies, and model scales. These findings provide valuable insights into the token-precision trade-off in KV cache compression. We plan to release our code in the near future.
Multi-view biomedical foundation models for molecule-target and property prediction
Foundation models applied to bio-molecular space hold promise to accelerate drug discovery. Molecular representation is key to building such models. Previous works have typically focused on a single representation or view of the molecules. Here, we develop a multi-view foundation model approach, that integrates molecular views of graph, image and text. Single-view foundation models are each pre-trained on a dataset of up to 200M molecules and then aggregated into combined representations. Our multi-view model is validated on a diverse set of 18 tasks, encompassing ligand-protein binding, molecular solubility, metabolism and toxicity. We show that the multi-view models perform robustly and are able to balance the strengths and weaknesses of specific views. We then apply this model to screen compounds against a large (>100 targets) set of G Protein-Coupled receptors (GPCRs). From this library of targets, we identify 33 that are related to Alzheimer's disease. On this subset, we employ our model to identify strong binders, which are validated through structure-based modeling and identification of key binding motifs.

MoRE: Multi-Modal Contrastive Pre-training with Transformers on X-Rays, ECGs, and Diagnostic Report
In this paper, we introduce a novel Multi-Modal Contrastive Pre-training Framework that synergistically combines X-rays, electrocardiograms (ECGs), and radiology/cardiology reports. Our approach leverages transformers to encode these diverse modalities into a unified representation space, aiming to enhance diagnostic accuracy and facilitate comprehensive patient assessments. We utilize LoRA-Peft to significantly reduce trainable parameters in the LLM and incorporate recent linear attention dropping strategy in the Vision Transformer(ViT) for smoother attention. Furthermore, we provide novel multimodal attention explanations and retrieval for our model. To the best of our knowledge, we are the first to propose an integrated model that combines X-ray, ECG, and Radiology/Cardiology Report with this approach. By utilizing contrastive loss, MoRE effectively aligns modality-specific features into a coherent embedding, which supports various downstream tasks such as zero-shot classification and multimodal retrieval. Employing our proposed methodology, we achieve state-of-the-art (SOTA) on the Mimic-IV, CheXpert, Edema Severity, and PtbXl downstream datasets, surpassing existing multimodal approaches. Our proposed framework shows significant improvements in capturing intricate inter-modal relationships and its robustness in medical diagnosis that establishes a framework for future research in multimodal learning in the healthcare sector.



Chain-of-Thoughts for Molecular Understanding
The adaptation of large language models (LLMs) to chemistry has shown promising performance in molecular understanding tasks, such as generating a text description from a molecule. However, proper reasoning based on molecular structural information remains a significant challenge, e.g., even advanced LLMs such as GPT-4o struggle to identify functional groups which are crucial for inferring the molecular property of interest. To address this limitation, we propose StructCoT, a structure-aware chain-of-thought (CoT) that enhances LLMs' understanding of molecular structures by explicitly injecting the key structural features of molecules. Moreover, we introduce two fine-tuning frameworks for adapting the existing LLMs to use our StructCoT. Our experiments demonstrate that incorporating StructCoT with our fine-tuning frameworks leads to consistent improvements in both molecular understanding tasks.

More Text, Less Point: Towards 3D Data-Efficient Point-Language Understanding
Enabling Large Language Models (LLMs) to comprehend the 3D physical world remains a significant challenge. Due to the lack of large-scale 3D-text pair datasets, the success of LLMs has yet to be replicated in 3D understanding. In this paper, we rethink this issue and propose a new task: 3D Data-Efficient Point-Language Understanding. The goal is to enable LLMs to achieve robust 3D object understanding with minimal 3D point cloud and text data pairs. To address this task, we introduce GreenPLM, which leverages more text data to compensate for the lack of 3D data. First, inspired by using CLIP to align images and text, we utilize a pre-trained point cloud-text encoder to map the 3D point cloud space to the text space. This mapping leaves us to seamlessly connect the text space with LLMs. Once the point-text-LLM connection is established, we further enhance text-LLM alignment by expanding the intermediate text space, thereby reducing the reliance on 3D point cloud data. Specifically, we generate 6M free-text descriptions of 3D objects, and design a three-stage training strategy to help LLMs better explore the intrinsic connections between different modalities. To achieve efficient modality alignment, we design a zero-parameter cross-attention module for token pooling. Extensive experimental results show that GreenPLM requires only 12% of the 3D training data used by existing state-of-the-art models to achieve superior 3D understanding. Remarkably, GreenPLM also achieves competitive performance using text-only data. The code and weights are available at: https://github.com/TangYuan96/GreenPLM.
Conan-embedding: General Text Embedding with More and Better Negative Samples
With the growing popularity of RAG, the capabilities of embedding models are gaining increasing attention. Embedding models are primarily trained through contrastive loss learning, with negative examples being a key component. Previous work has proposed various hard negative mining strategies, but these strategies are typically employed as preprocessing steps. In this paper, we propose the conan-embedding model, which maximizes the utilization of more and higher-quality negative examples. Specifically, since the model's ability to handle preprocessed negative examples evolves during training, we propose dynamic hard negative mining method to expose the model to more challenging negative examples throughout the training process. Secondly, contrastive learning requires as many negative examples as possible but is limited by GPU memory constraints. Therefore, we use a Cross-GPU balancing Loss to provide more negative examples for embedding training and balance the batch size across multiple tasks. Moreover, we also discovered that the prompt-response pairs from LLMs can be used for embedding training. Our approach effectively enhances the capabilities of embedding models, currently ranking first on the Chinese leaderboard of Massive text embedding benchmark
Making Task-Oriented Dialogue Datasets More Natural by Synthetically Generating Indirect User Requests
Indirect User Requests (IURs), such as "It's cold in here" instead of "Could you please increase the temperature?" are common in human-human task-oriented dialogue and require world knowledge and pragmatic reasoning from the listener. While large language models (LLMs) can handle these requests effectively, smaller models deployed on virtual assistants often struggle due to resource constraints. Moreover, existing task-oriented dialogue benchmarks lack sufficient examples of complex discourse phenomena such as indirectness. To address this, we propose a set of linguistic criteria along with an LLM-based pipeline for generating realistic IURs to test natural language understanding (NLU) and dialogue state tracking (DST) models before deployment in a new domain. We also release IndirectRequests, a dataset of IURs based on the Schema Guided Dialog (SGD) corpus, as a comparative testbed for evaluating the performance of smaller models in handling indirect requests.
Get more for less: Principled Data Selection for Warming Up Fine-Tuning in LLMs
This work focuses on leveraging and selecting from vast, unlabeled, open data to pre-fine-tune a pre-trained language model. The goal is to minimize the need for costly domain-specific data for subsequent fine-tuning while achieving desired performance levels. While many data selection algorithms have been designed for small-scale applications, rendering them unsuitable for our context, some emerging methods do cater to language data scales. However, they often prioritize data that aligns with the target distribution. While this strategy may be effective when training a model from scratch, it can yield limited results when the model has already been pre-trained on a different distribution. Differing from prior work, our key idea is to select data that nudges the pre-training distribution closer to the target distribution. We show the optimality of this approach for fine-tuning tasks under certain conditions. We demonstrate the efficacy of our methodology across a diverse array of tasks (NLU, NLG, zero-shot) with models up to 2.7B, showing that it consistently surpasses other selection methods. Moreover, our proposed method is significantly faster than existing techniques, scaling to millions of samples within a single GPU hour. Our code is open-sourced (Code repository: https://anonymous.4open.science/r/DV4LLM-D761/ ). While fine-tuning offers significant potential for enhancing performance across diverse tasks, its associated costs often limit its widespread adoption; with this work, we hope to lay the groundwork for cost-effective fine-tuning, making its benefits more accessible.

MoReVQA: Exploring Modular Reasoning Models for Video Question Answering
This paper addresses the task of video question answering (videoQA) via a decomposed multi-stage, modular reasoning framework. Previous modular methods have shown promise with a single planning stage ungrounded in visual content. However, through a simple and effective baseline, we find that such systems can lead to brittle behavior in practice for challenging videoQA settings. Thus, unlike traditional single-stage planning methods, we propose a multi-stage system consisting of an event parser, a grounding stage, and a final reasoning stage in conjunction with an external memory. All stages are training-free, and performed using few-shot prompting of large models, creating interpretable intermediate outputs at each stage. By decomposing the underlying planning and task complexity, our method, MoReVQA, improves over prior work on standard videoQA benchmarks (NExT-QA, iVQA, EgoSchema, ActivityNet-QA) with state-of-the-art results, and extensions to related tasks (grounded videoQA, paragraph captioning).
MoleculeQA: A Dataset to Evaluate Factual Accuracy in Molecular Comprehension
Large language models are playing an increasingly significant role in molecular research, yet existing models often generate erroneous information, posing challenges to accurate molecular comprehension. Traditional evaluation metrics for generated content fail to assess a model's accuracy in molecular understanding. To rectify the absence of factual evaluation, we present MoleculeQA, a novel question answering (QA) dataset which possesses 62K QA pairs over 23K molecules. Each QA pair, composed of a manual question, a positive option and three negative options, has consistent semantics with a molecular description from authoritative molecular corpus. MoleculeQA is not only the first benchmark for molecular factual bias evaluation but also the largest QA dataset for molecular research. A comprehensive evaluation on MoleculeQA for existing molecular LLMs exposes their deficiencies in specific areas and pinpoints several particularly crucial factors for molecular understanding.
DecompOpt: Controllable and Decomposed Diffusion Models for Structure-based Molecular Optimization
Recently, 3D generative models have shown promising performances in structure-based drug design by learning to generate ligands given target binding sites. However, only modeling the target-ligand distribution can hardly fulfill one of the main goals in drug discovery -- designing novel ligands with desired properties, e.g., high binding affinity, easily synthesizable, etc. This challenge becomes particularly pronounced when the target-ligand pairs used for training do not align with these desired properties. Moreover, most existing methods aim at solving de novo design task, while many generative scenarios requiring flexible controllability, such as R-group optimization and scaffold hopping, have received little attention. In this work, we propose DecompOpt, a structure-based molecular optimization method based on a controllable and decomposed diffusion model. DecompOpt presents a new generation paradigm which combines optimization with conditional diffusion models to achieve desired properties while adhering to the molecular grammar. Additionally, DecompOpt offers a unified framework covering both de novo design and controllable generation. To achieve so, ligands are decomposed into substructures which allows fine-grained control and local optimization. Experiments show that DecompOpt can efficiently generate molecules with improved properties than strong de novo baselines, and demonstrate great potential in controllable generation tasks.

MORE: Multi-mOdal REtrieval Augmented Generative Commonsense Reasoning
Since commonsense information has been recorded significantly less frequently than its existence, language models pre-trained by text generation have difficulty to learn sufficient commonsense knowledge. Several studies have leveraged text retrieval to augment the models' commonsense ability. Unlike text, images capture commonsense information inherently but little effort has been paid to effectively utilize them. In this work, we propose a novel Multi-mOdal REtrieval (MORE) augmentation framework, to leverage both text and images to enhance the commonsense ability of language models. Extensive experiments on the Common-Gen task have demonstrated the efficacy of MORE based on the pre-trained models of both single and multiple modalities.
Soft Prompt Tuning for Cross-Lingual Transfer: When Less is More
Soft Prompt Tuning (SPT) is a parameter-efficient method for adapting pre-trained language models (PLMs) to specific tasks by inserting learnable embeddings, or soft prompts, at the input layer of the PLM, without modifying its parameters. This paper investigates the potential of SPT for cross-lingual transfer. Unlike previous studies on SPT for cross-lingual transfer that often fine-tune both the soft prompt and the model parameters, we adhere to the original intent of SPT by keeping the model parameters frozen and only training the soft prompt. This does not only reduce the computational cost and storage overhead of full-model fine-tuning, but we also demonstrate that this very parameter efficiency intrinsic to SPT can enhance cross-lingual transfer performance to linguistically distant languages. Moreover, we explore how different factors related to the prompt, such as the length or its reparameterization, affect cross-lingual transfer performance.


More is Better in Modern Machine Learning: when Infinite Overparameterization is Optimal and Overfitting is Obligatory
In our era of enormous neural networks, empirical progress has been driven by the philosophy that more is better. Recent deep learning practice has found repeatedly that larger model size, more data, and more computation (resulting in lower training loss) improves performance. In this paper, we give theoretical backing to these empirical observations by showing that these three properties hold in random feature (RF) regression, a class of models equivalent to shallow networks with only the last layer trained. Concretely, we first show that the test risk of RF regression decreases monotonically with both the number of features and the number of samples, provided the ridge penalty is tuned optimally. In particular, this implies that infinite width RF architectures are preferable to those of any finite width. We then proceed to demonstrate that, for a large class of tasks characterized by powerlaw eigenstructure, training to near-zero training loss is obligatory: near-optimal performance can only be achieved when the training error is much smaller than the test error. Grounding our theory in real-world data, we find empirically that standard computer vision tasks with convolutional neural tangent kernels clearly fall into this class. Taken together, our results tell a simple, testable story of the benefits of overparameterization, overfitting, and more data in random feature models.

More Samples or More Prompts? Exploring Effective In-Context Sampling for LLM Few-Shot Prompt Engineering
While most existing works on LLM prompting techniques focus only on how to select a better set of data samples inside one single prompt input (In-Context Learning or ICL), why can not we design and leverage multiple prompts together to further improve the LLM's performance? In this work, we propose In-Context Sampling (ICS), a low-resource LLM prompting technique to produce confident predictions by optimizing the construction of multiple ICL prompt inputs. Extensive experiments with three open-source LLMs (FlanT5-XL, Mistral-7B, and Mixtral-8x7B) on four NLI datasets (e-SNLI, Multi-NLI, ANLI, and Contract-NLI) and one QA dataset (CommonsenseQA) illustrate that ICS can consistently enhance LLMs' performance. An in-depth evaluation with three data similarity-based ICS strategies suggests that these strategies can further elevate LLM's performance, which sheds light on a new yet promising future research direction.

A molecular Ferroelectric thin film of imidazolium perchlorate on Silicon
Molecular ferroelectric materials have attracted widespread attention due to their abundant chemical diversity, structural tunability, low synthesis temperature, and high flexibility. Meanwhile, the integration of molecular ferroelectric materials and Si is still challenging, while the fundamental understanding of the ferroelectric switching process is still lacking. Herein, we have successfully synthesized the imidazole perchlorate (ImClO4) single crystals and a series of high-quality highly-oriented thin films on a Si substrate. A high inverse piezoelectric coefficient (55.7 pm/V) is demonstrated for the thin films. Two types of domain bands can be observed (in the size of a few microns): type-I band tilts ~60{\deg} with respect to the horizontal axis, while the type-II band is perpendicular to the horizontal axis. Most of the domain walls (DWs) are 180{\deg} DWs for the two bands, while some 109{\deg} DWs can also be observed. Interestingly, the DWs in type-I band are curved, charged domain walls; while the 180{\deg} DWs in type-II band are straight, noncharged domain walls. After applying +20 V for 5 s through a PFM tip, the 180{\deg} DWs in type-I band shrink first, then disconnect from the band boundary, forming a needle-like domain with a size of ~100 nm. The needle-like domain will extend toward the band boundary after an inverse bias is applied (-20 V), and expand along the band boundary after touching the boundary. Whereas for the type-II domain band, the 180{\deg} DWs are more mobile than the 109{\deg} domain walls, which displaces ~500 nm after applying +20 V. While such displacement is much shorter after the application of a negative bias for the same duration, starting from the positively poled sample. We hope to spur further interest in the on-chip design of the molecular ferroelectrics based electronic devices.
Learning Over Molecular Conformer Ensembles: Datasets and Benchmarks
Molecular Representation Learning (MRL) has proven impactful in numerous biochemical applications such as drug discovery and enzyme design. While Graph Neural Networks (GNNs) are effective at learning molecular representations from a 2D molecular graph or a single 3D structure, existing works often overlook the flexible nature of molecules, which continuously interconvert across conformations via chemical bond rotations and minor vibrational perturbations. To better account for molecular flexibility, some recent works formulate MRL as an ensemble learning problem, focusing on explicitly learning from a set of conformer structures. However, most of these studies have limited datasets, tasks, and models. In this work, we introduce the first MoleculAR Conformer Ensemble Learning (MARCEL) benchmark to thoroughly evaluate the potential of learning on conformer ensembles and suggest promising research directions. MARCEL includes four datasets covering diverse molecule- and reaction-level properties of chemically diverse molecules including organocatalysts and transition-metal catalysts, extending beyond the scope of common GNN benchmarks that are confined to drug-like molecules. In addition, we conduct a comprehensive empirical study, which benchmarks representative 1D, 2D, and 3D molecular representation learning models, along with two strategies that explicitly incorporate conformer ensembles into 3D MRL models. Our findings reveal that direct learning from an accessible conformer space can improve performance on a variety of tasks and models.




More complex encoder is not all you need
U-Net and its variants have been widely used in medical image segmentation. However, most current U-Net variants confine their improvement strategies to building more complex encoder, while leaving the decoder unchanged or adopting a simple symmetric structure. These approaches overlook the true functionality of the decoder: receiving low-resolution feature maps from the encoder and restoring feature map resolution and lost information through upsampling. As a result, the decoder, especially its upsampling component, plays a crucial role in enhancing segmentation outcomes. However, in 3D medical image segmentation, the commonly used transposed convolution can result in visual artifacts. This issue stems from the absence of direct relationship between adjacent pixels in the output feature map. Furthermore, plain encoder has already possessed sufficient feature extraction capability because downsampling operation leads to the gradual expansion of the receptive field, but the loss of information during downsampling process is unignorable. To address the gap in relevant research, we extend our focus beyond the encoder and introduce neU-Net (i.e., not complex encoder U-Net), which incorporates a novel Sub-pixel Convolution for upsampling to construct a powerful decoder. Additionally, we introduce multi-scale wavelet inputs module on the encoder side to provide additional information. Our model design achieves excellent results, surpassing other state-of-the-art methods on both the Synapse and ACDC datasets.
From Artificially Real to Real: Leveraging Pseudo Data from Large Language Models for Low-Resource Molecule Discovery
Molecule discovery serves as a cornerstone in numerous scientific domains, fueling the development of new materials and innovative drug designs. Recent developments of in-silico molecule discovery have highlighted the promising results of cross-modal techniques, which bridge molecular structures with their descriptive annotations. However, these cross-modal methods frequently encounter the issue of data scarcity, hampering their performance and application. In this paper, we address the low-resource challenge by utilizing artificially-real data generated by Large Language Models (LLMs). We first introduce a retrieval-based prompting strategy to construct high-quality pseudo data, then explore the optimal method to effectively leverage this pseudo data. Experiments show that using pseudo data for domain adaptation outperforms all existing methods, while also requiring a smaller model scale, reduced data size and lower training cost, highlighting its efficiency. Furthermore, our method shows a sustained improvement as the volume of pseudo data increases, revealing the great potential of pseudo data in advancing low-resource cross-modal molecule discovery.
Can Large Language Models Empower Molecular Property Prediction?
Molecular property prediction has gained significant attention due to its transformative potential in multiple scientific disciplines. Conventionally, a molecule graph can be represented either as a graph-structured data or a SMILES text. Recently, the rapid development of Large Language Models (LLMs) has revolutionized the field of NLP. Although it is natural to utilize LLMs to assist in understanding molecules represented by SMILES, the exploration of how LLMs will impact molecular property prediction is still in its early stage. In this work, we advance towards this objective through two perspectives: zero/few-shot molecular classification, and using the new explanations generated by LLMs as representations of molecules. To be specific, we first prompt LLMs to do in-context molecular classification and evaluate their performance. After that, we employ LLMs to generate semantically enriched explanations for the original SMILES and then leverage that to fine-tune a small-scale LM model for multiple downstream tasks. The experimental results highlight the superiority of text explanations as molecular representations across multiple benchmark datasets, and confirm the immense potential of LLMs in molecular property prediction tasks. Codes are available at https://github.com/ChnQ/LLM4Mol.
Machine-learned molecular mechanics force field for the simulation of protein-ligand systems and beyond
The development of reliable and extensible molecular mechanics (MM) force fields -- fast, empirical models characterizing the potential energy surface of molecular systems -- is indispensable for biomolecular simulation and computer-aided drug design. Here, we introduce a generalized and extensible machine-learned MM force field, espaloma-0.3, and an end-to-end differentiable framework using graph neural networks to overcome the limitations of traditional rule-based methods. Trained in a single GPU-day to fit a large and diverse quantum chemical dataset of over 1.1M energy and force calculations, espaloma-0.3 reproduces quantum chemical energetic properties of chemical domains highly relevant to drug discovery, including small molecules, peptides, and nucleic acids. Moreover, this force field maintains the quantum chemical energy-minimized geometries of small molecules and preserves the condensed phase properties of peptides, self-consistently parametrizing proteins and ligands to produce stable simulations leading to highly accurate predictions of binding free energies. This methodology demonstrates significant promise as a path forward for systematically building more accurate force fields that are easily extensible to new chemical domains of interest.


Von Mises Mixture Distributions for Molecular Conformation Generation
Molecules are frequently represented as graphs, but the underlying 3D molecular geometry (the locations of the atoms) ultimately determines most molecular properties. However, most molecules are not static and at room temperature adopt a wide variety of geometries or conformations. The resulting distribution on geometries p(x) is known as the Boltzmann distribution, and many molecular properties are expectations computed under this distribution. Generating accurate samples from the Boltzmann distribution is therefore essential for computing these expectations accurately. Traditional sampling-based methods are computationally expensive, and most recent machine learning-based methods have focused on identifying modes in this distribution rather than generating true samples. Generating such samples requires capturing conformational variability, and it has been widely recognized that the majority of conformational variability in molecules arises from rotatable bonds. In this work, we present VonMisesNet, a new graph neural network that captures conformational variability via a variational approximation of rotatable bond torsion angles as a mixture of von Mises distributions. We demonstrate that VonMisesNet can generate conformations for arbitrary molecules in a way that is both physically accurate with respect to the Boltzmann distribution and orders of magnitude faster than existing sampling methods.
TensorNet: Cartesian Tensor Representations for Efficient Learning of Molecular Potentials
The development of efficient machine learning models for molecular systems representation is becoming crucial in scientific research. We introduce TensorNet, an innovative O(3)-equivariant message-passing neural network architecture that leverages Cartesian tensor representations. By using Cartesian tensor atomic embeddings, feature mixing is simplified through matrix product operations. Furthermore, the cost-effective decomposition of these tensors into rotation group irreducible representations allows for the separate processing of scalars, vectors, and tensors when necessary. Compared to higher-rank spherical tensor models, TensorNet demonstrates state-of-the-art performance with significantly fewer parameters. For small molecule potential energies, this can be achieved even with a single interaction layer. As a result of all these properties, the model's computational cost is substantially decreased. Moreover, the accurate prediction of vector and tensor molecular quantities on top of potential energies and forces is possible. In summary, TensorNet's framework opens up a new space for the design of state-of-the-art equivariant models.
A Group Symmetric Stochastic Differential Equation Model for Molecule Multi-modal Pretraining
Molecule pretraining has quickly become the go-to schema to boost the performance of AI-based drug discovery. Naturally, molecules can be represented as 2D topological graphs or 3D geometric point clouds. Although most existing pertaining methods focus on merely the single modality, recent research has shown that maximizing the mutual information (MI) between such two modalities enhances the molecule representation ability. Meanwhile, existing molecule multi-modal pretraining approaches approximate MI based on the representation space encoded from the topology and geometry, thus resulting in the loss of critical structural information of molecules. To address this issue, we propose MoleculeSDE. MoleculeSDE leverages group symmetric (e.g., SE(3)-equivariant and reflection-antisymmetric) stochastic differential equation models to generate the 3D geometries from 2D topologies, and vice versa, directly in the input space. It not only obtains tighter MI bound but also enables prosperous downstream tasks than the previous work. By comparing with 17 pretraining baselines, we empirically verify that MoleculeSDE can learn an expressive representation with state-of-the-art performance on 26 out of 32 downstream tasks.
Dreams Are More "Predictable'' Than You Think
A consistent body of evidence suggests that dream reports significantly vary from other types of textual transcripts with respect to semantic content. Furthermore, it appears to be a widespread belief in the dream/sleep research community that dream reports constitute rather ``unique'' strings of text. This might be a notable issue for the growing amount of approaches using natural language processing (NLP) tools to automatically analyse dream reports, as they largely rely on neural models trained on non-dream corpora scraped from the web. In this work, I will adopt state-of-the-art (SotA) large language models (LLMs), to study if and how dream reports deviate from other human-generated text strings, such as Wikipedia. Results show that, taken as a whole, DreamBank does not deviate from Wikipedia. Moreover, on average, single dream reports are significantly more predictable than Wikipedia articles. Preliminary evidence suggests that word count, gender, and visual impairment can significantly shape how predictable a dream report can appear to the model.

Conditional Graph Information Bottleneck for Molecular Relational Learning
Molecular relational learning, whose goal is to learn the interaction behavior between molecular pairs, got a surge of interest in molecular sciences due to its wide range of applications. Recently, graph neural networks have recently shown great success in molecular relational learning by modeling a molecule as a graph structure, and considering atom-level interactions between two molecules. Despite their success, existing molecular relational learning methods tend to overlook the nature of chemistry, i.e., a chemical compound is composed of multiple substructures such as functional groups that cause distinctive chemical reactions. In this work, we propose a novel relational learning framework, called CGIB, that predicts the interaction behavior between a pair of graphs by detecting core subgraphs therein. The main idea is, given a pair of graphs, to find a subgraph from a graph that contains the minimal sufficient information regarding the task at hand conditioned on the paired graph based on the principle of conditional graph information bottleneck. We argue that our proposed method mimics the nature of chemical reactions, i.e., the core substructure of a molecule varies depending on which other molecule it interacts with. Extensive experiments on various tasks with real-world datasets demonstrate the superiority of CGIB over state-of-the-art baselines. Our code is available at https://github.com/Namkyeong/CGIB.
Timewarp: Transferable Acceleration of Molecular Dynamics by Learning Time-Coarsened Dynamics
Molecular dynamics (MD) simulation is a widely used technique to simulate molecular systems, most commonly at the all-atom resolution where equations of motion are integrated with timesteps on the order of femtoseconds (1fs=10^{-15}s). MD is often used to compute equilibrium properties, which requires sampling from an equilibrium distribution such as the Boltzmann distribution. However, many important processes, such as binding and folding, occur over timescales of milliseconds or beyond, and cannot be efficiently sampled with conventional MD. Furthermore, new MD simulations need to be performed for each molecular system studied. We present Timewarp, an enhanced sampling method which uses a normalising flow as a proposal distribution in a Markov chain Monte Carlo method targeting the Boltzmann distribution. The flow is trained offline on MD trajectories and learns to make large steps in time, simulating the molecular dynamics of 10^{5} - 10^{6}:fs. Crucially, Timewarp is transferable between molecular systems: once trained, we show that it generalises to unseen small peptides (2-4 amino acids) at all-atom resolution, exploring their metastable states and providing wall-clock acceleration of sampling compared to standard MD. Our method constitutes an important step towards general, transferable algorithms for accelerating MD.
MoreauGrad: Sparse and Robust Interpretation of Neural Networks via Moreau Envelope
Explaining the predictions of deep neural nets has been a topic of great interest in the computer vision literature. While several gradient-based interpretation schemes have been proposed to reveal the influential variables in a neural net's prediction, standard gradient-based interpretation frameworks have been commonly observed to lack robustness to input perturbations and flexibility for incorporating prior knowledge of sparsity and group-sparsity structures. In this work, we propose MoreauGrad as an interpretation scheme based on the classifier neural net's Moreau envelope. We demonstrate that MoreauGrad results in a smooth and robust interpretation of a multi-layer neural network and can be efficiently computed through first-order optimization methods. Furthermore, we show that MoreauGrad can be naturally combined with L_1-norm regularization techniques to output a sparse or group-sparse explanation which are prior conditions applicable to a wide range of deep learning applications. We empirically evaluate the proposed MoreauGrad scheme on standard computer vision datasets, showing the qualitative and quantitative success of the MoreauGrad approach in comparison to standard gradient-based interpretation methods.
Multi-modal Molecule Structure-text Model for Text-based Retrieval and Editing
There is increasing adoption of artificial intelligence in drug discovery. However, existing studies use machine learning to mainly utilize the chemical structures of molecules but ignore the vast textual knowledge available in chemistry. Incorporating textual knowledge enables us to realize new drug design objectives, adapt to text-based instructions and predict complex biological activities. Here we present a multi-modal molecule structure-text model, MoleculeSTM, by jointly learning molecules' chemical structures and textual descriptions via a contrastive learning strategy. To train MoleculeSTM, we construct a large multi-modal dataset, namely, PubChemSTM, with over 280,000 chemical structure-text pairs. To demonstrate the effectiveness and utility of MoleculeSTM, we design two challenging zero-shot tasks based on text instructions, including structure-text retrieval and molecule editing. MoleculeSTM has two main properties: open vocabulary and compositionality via natural language. In experiments, MoleculeSTM obtains the state-of-the-art generalization ability to novel biochemical concepts across various benchmarks.
Hybrid Quantum Generative Adversarial Networks for Molecular Simulation and Drug Discovery
In molecular research, simulation \& design of molecules are key areas with significant implications for drug development, material science, and other fields. Current classical computational power falls inadequate to simulate any more than small molecules, let alone protein chains on hundreds of peptide. Therefore these experiment are done physically in wet-lab, but it takes a lot of time \& not possible to examine every molecule due to the size of the search area, tens of billions of dollars are spent every year in these research experiments. Molecule simulation \& design has lately advanced significantly by machine learning models, A fresh perspective on the issue of chemical synthesis is provided by deep generative models for graph-structured data. By optimising differentiable models that produce molecular graphs directly, it is feasible to avoid costly search techniques in the discrete and huge space of chemical structures. But these models also suffer from computational limitations when dimensions become huge and consume huge amount of resources. Quantum Generative machine learning in recent years have shown some empirical results promising significant advantages over classical counterparts.
A Graph Is More Than Its Nodes: Towards Structured Uncertainty-Aware Learning on Graphs
Current graph neural networks (GNNs) that tackle node classification on graphs tend to only focus on nodewise scores and are solely evaluated by nodewise metrics. This limits uncertainty estimation on graphs since nodewise marginals do not fully characterize the joint distribution given the graph structure. In this work, we propose novel edgewise metrics, namely the edgewise expected calibration error (ECE) and the agree/disagree ECEs, which provide criteria for uncertainty estimation on graphs beyond the nodewise setting. Our experiments demonstrate that the proposed edgewise metrics can complement the nodewise results and yield additional insights. Moreover, we show that GNN models which consider the structured prediction problem on graphs tend to have better uncertainty estimations, which illustrates the benefit of going beyond the nodewise setting.

MolScribe: Robust Molecular Structure Recognition with Image-To-Graph Generation
Molecular structure recognition is the task of translating a molecular image into its graph structure. Significant variation in drawing styles and conventions exhibited in chemical literature poses a significant challenge for automating this task. In this paper, we propose MolScribe, a novel image-to-graph generation model that explicitly predicts atoms and bonds, along with their geometric layouts, to construct the molecular structure. Our model flexibly incorporates symbolic chemistry constraints to recognize chirality and expand abbreviated structures. We further develop data augmentation strategies to enhance the model robustness against domain shifts. In experiments on both synthetic and realistic molecular images, MolScribe significantly outperforms previous models, achieving 76-93% accuracy on public benchmarks. Chemists can also easily verify MolScribe's prediction, informed by its confidence estimation and atom-level alignment with the input image. MolScribe is publicly available through Python and web interfaces: https://github.com/thomas0809/MolScribe.
Knowledge-informed Molecular Learning: A Survey on Paradigm Transfer
Machine learning, notably deep learning, has significantly propelled molecular investigations within the biochemical sphere. Traditionally, modeling for such research has centered around a handful of paradigms. For instance, the prediction paradigm is frequently deployed for tasks such as molecular property prediction. To enhance the generation and decipherability of purely data-driven models, scholars have integrated biochemical domain knowledge into these molecular study models. This integration has sparked a surge in paradigm transfer, which is solving one molecular learning task by reformulating it as another one. With the emergence of Large Language Models, these paradigms have demonstrated an escalating trend towards harmonized unification. In this work, we delineate a literature survey focused on knowledge-informed molecular learning from the perspective of paradigm transfer. We classify the paradigms, scrutinize their methodologies, and dissect the contribution of domain knowledge. Moreover, we encapsulate prevailing trends and identify intriguing avenues for future exploration in molecular learning.
Vision Models Are More Robust And Fair When Pretrained On Uncurated Images Without Supervision
Discriminative self-supervised learning allows training models on any random group of internet images, and possibly recover salient information that helps differentiate between the images. Applied to ImageNet, this leads to object centric features that perform on par with supervised features on most object-centric downstream tasks. In this work, we question if using this ability, we can learn any salient and more representative information present in diverse unbounded set of images from across the globe. To do so, we train models on billions of random images without any data pre-processing or prior assumptions about what we want the model to learn. We scale our model size to dense 10 billion parameters to avoid underfitting on a large data size. We extensively study and validate our model performance on over 50 benchmarks including fairness, robustness to distribution shift, geographical diversity, fine grained recognition, image copy detection and many image classification datasets. The resulting model, not only captures well semantic information, it also captures information about artistic style and learns salient information such as geolocations and multilingual word embeddings based on visual content only. More importantly, we discover that such model is more robust, more fair, less harmful and less biased than supervised models or models trained on object centric datasets such as ImageNet.
More Control for Free! Image Synthesis with Semantic Diffusion Guidance
Controllable image synthesis models allow creation of diverse images based on text instructions or guidance from a reference image. Recently, denoising diffusion probabilistic models have been shown to generate more realistic imagery than prior methods, and have been successfully demonstrated in unconditional and class-conditional settings. We investigate fine-grained, continuous control of this model class, and introduce a novel unified framework for semantic diffusion guidance, which allows either language or image guidance, or both. Guidance is injected into a pretrained unconditional diffusion model using the gradient of image-text or image matching scores, without re-training the diffusion model. We explore CLIP-based language guidance as well as both content and style-based image guidance in a unified framework. Our text-guided synthesis approach can be applied to datasets without associated text annotations. We conduct experiments on FFHQ and LSUN datasets, and show results on fine-grained text-guided image synthesis, synthesis of images related to a style or content reference image, and examples with both textual and image guidance.

Molecular Contrastive Learning with Chemical Element Knowledge Graph
Molecular representation learning contributes to multiple downstream tasks such as molecular property prediction and drug design. To properly represent molecules, graph contrastive learning is a promising paradigm as it utilizes self-supervision signals and has no requirements for human annotations. However, prior works fail to incorporate fundamental domain knowledge into graph semantics and thus ignore the correlations between atoms that have common attributes but are not directly connected by bonds. To address these issues, we construct a Chemical Element Knowledge Graph (KG) to summarize microscopic associations between elements and propose a novel Knowledge-enhanced Contrastive Learning (KCL) framework for molecular representation learning. KCL framework consists of three modules. The first module, knowledge-guided graph augmentation, augments the original molecular graph based on the Chemical Element KG. The second module, knowledge-aware graph representation, extracts molecular representations with a common graph encoder for the original molecular graph and a Knowledge-aware Message Passing Neural Network (KMPNN) to encode complex information in the augmented molecular graph. The final module is a contrastive objective, where we maximize agreement between these two views of molecular graphs. Extensive experiments demonstrated that KCL obtained superior performances against state-of-the-art baselines on eight molecular datasets. Visualization experiments properly interpret what KCL has learned from atoms and attributes in the augmented molecular graphs. Our codes and data are available at https://github.com/ZJU-Fangyin/KCL.
Molecular Graph Generation via Geometric Scattering
Graph neural networks (GNNs) have been used extensively for addressing problems in drug design and discovery. Both ligand and target molecules are represented as graphs with node and edge features encoding information about atomic elements and bonds respectively. Although existing deep learning models perform remarkably well at predicting physicochemical properties and binding affinities, the generation of new molecules with optimized properties remains challenging. Inherently, most GNNs perform poorly in whole-graph representation due to the limitations of the message-passing paradigm. Furthermore, step-by-step graph generation frameworks that use reinforcement learning or other sequential processing can be slow and result in a high proportion of invalid molecules with substantial post-processing needed in order to satisfy the principles of stoichiometry. To address these issues, we propose a representation-first approach to molecular graph generation. We guide the latent representation of an autoencoder by capturing graph structure information with the geometric scattering transform and apply penalties that structure the representation also by molecular properties. We show that this highly structured latent space can be directly used for molecular graph generation by the use of a GAN. We demonstrate that our architecture learns meaningful representations of drug datasets and provides a platform for goal-directed drug synthesis.

Molecule3D: A Benchmark for Predicting 3D Geometries from Molecular Graphs
Graph neural networks are emerging as promising methods for modeling molecular graphs, in which nodes and edges correspond to atoms and chemical bonds, respectively. Recent studies show that when 3D molecular geometries, such as bond lengths and angles, are available, molecular property prediction tasks can be made more accurate. However, computing of 3D molecular geometries requires quantum calculations that are computationally prohibitive. For example, accurate calculation of 3D geometries of a small molecule requires hours of computing time using density functional theory (DFT). Here, we propose to predict the ground-state 3D geometries from molecular graphs using machine learning methods. To make this feasible, we develop a benchmark, known as Molecule3D, that includes a dataset with precise ground-state geometries of approximately 4 million molecules derived from DFT. We also provide a set of software tools for data processing, splitting, training, and evaluation, etc. Specifically, we propose to assess the error and validity of predicted geometries using four metrics. We implement two baseline methods that either predict the pairwise distance between atoms or atom coordinates in 3D space. Experimental results show that, compared with generating 3D geometries with RDKit, our method can achieve comparable prediction accuracy but with much smaller computational costs. Our Molecule3D is available as a module of the MoleculeX software library (https://github.com/divelab/MoleculeX).

More than Encoder: Introducing Transformer Decoder to Upsample
Medical image segmentation methods downsample images for feature extraction and then upsample them to restore resolution for pixel-level predictions. In such a schema, upsample technique is vital in restoring information for better performance. However, existing upsample techniques leverage little information from downsampling paths. The local and detailed feature from the shallower layer such as boundary and tissue texture is particularly more important in medical segmentation compared with natural image segmentation. To this end, we propose a novel upsample approach for medical image segmentation, Window Attention Upsample (WAU), which upsamples features conditioned on local and detailed features from downsampling path in local windows by introducing attention decoders of Transformer. WAU could serve as a general upsample method and be incorporated into any segmentation model that possesses lateral connections. We first propose the Attention Upsample which consists of Attention Decoder (AD) and bilinear upsample. AD leverages pixel-level attention to model long-range dependency and global information for a better upsample. Bilinear upsample is introduced as the residual connection to complement the upsampled features. Moreover, considering the extensive memory and computation cost of pixel-level attention, we further design a window attention scheme to restrict attention computation in local windows instead of the global range. We evaluate our method (WAU) on classic U-Net structure with lateral connections and achieve state-of-the-art performance on Synapse multi-organ segmentation, Medical Segmentation Decathlon (MSD) Brain, and Automatic Cardiac Diagnosis Challenge (ACDC) datasets. We also validate the effectiveness of our method on multiple classic architectures and achieve consistent improvement.
Making Attention Mechanisms More Robust and Interpretable with Virtual Adversarial Training
Although attention mechanisms have become fundamental components of deep learning models, they are vulnerable to perturbations, which may degrade the prediction performance and model interpretability. Adversarial training (AT) for attention mechanisms has successfully reduced such drawbacks by considering adversarial perturbations. However, this technique requires label information, and thus, its use is limited to supervised settings. In this study, we explore the concept of incorporating virtual AT (VAT) into the attention mechanisms, by which adversarial perturbations can be computed even from unlabeled data. To realize this approach, we propose two general training techniques, namely VAT for attention mechanisms (Attention VAT) and "interpretable" VAT for attention mechanisms (Attention iVAT), which extend AT for attention mechanisms to a semi-supervised setting. In particular, Attention iVAT focuses on the differences in attention; thus, it can efficiently learn clearer attention and improve model interpretability, even with unlabeled data. Empirical experiments based on six public datasets revealed that our techniques provide better prediction performance than conventional AT-based as well as VAT-based techniques, and stronger agreement with evidence that is provided by humans in detecting important words in sentences. Moreover, our proposal offers these advantages without needing to add the careful selection of unlabeled data. That is, even if the model using our VAT-based technique is trained on unlabeled data from a source other than the target task, both the prediction performance and model interpretability can be improved.

More Photos are All You Need: Semi-Supervised Learning for Fine-Grained Sketch Based Image Retrieval
A fundamental challenge faced by existing Fine-Grained Sketch-Based Image Retrieval (FG-SBIR) models is the data scarcity -- model performances are largely bottlenecked by the lack of sketch-photo pairs. Whilst the number of photos can be easily scaled, each corresponding sketch still needs to be individually produced. In this paper, we aim to mitigate such an upper-bound on sketch data, and study whether unlabelled photos alone (of which they are many) can be cultivated for performances gain. In particular, we introduce a novel semi-supervised framework for cross-modal retrieval that can additionally leverage large-scale unlabelled photos to account for data scarcity. At the centre of our semi-supervision design is a sequential photo-to-sketch generation model that aims to generate paired sketches for unlabelled photos. Importantly, we further introduce a discriminator guided mechanism to guide against unfaithful generation, together with a distillation loss based regularizer to provide tolerance against noisy training samples. Last but not least, we treat generation and retrieval as two conjugate problems, where a joint learning procedure is devised for each module to mutually benefit from each other. Extensive experiments show that our semi-supervised model yields significant performance boost over the state-of-the-art supervised alternatives, as well as existing methods that can exploit unlabelled photos for FG-SBIR.

Moroccan Dialect -Darija- Open Dataset
Darija Open Dataset (DODa) is an open-source project for the Moroccan dialect. With more than 10,000 entries DODa is arguably the largest open-source collaborative project for Darija-English translation built for Natural Language Processing purposes. In fact, besides semantic categorization, DODa also adopts a syntactic one, presents words under different spellings, offers verb-to-noun and masculine-to-feminine correspondences, contains the conjugation of hundreds of verbs in different tenses, and many other subsets to help researchers better understand and study Moroccan dialect. This data paper presents a description of DODa, its features, how it was collected, as well as a first application in Image Classification using ImageNet labels translated to Darija. This collaborative project is hosted on GitHub platform under MIT's Open-Source license and aims to be a standard resource for researchers, students, and anyone who is interested in Moroccan Dialect
SMILES Transformer: Pre-trained Molecular Fingerprint for Low Data Drug Discovery
In drug-discovery-related tasks such as virtual screening, machine learning is emerging as a promising way to predict molecular properties. Conventionally, molecular fingerprints (numerical representations of molecules) are calculated through rule-based algorithms that map molecules to a sparse discrete space. However, these algorithms perform poorly for shallow prediction models or small datasets. To address this issue, we present SMILES Transformer. Inspired by Transformer and pre-trained language models from natural language processing, SMILES Transformer learns molecular fingerprints through unsupervised pre-training of the sequence-to-sequence language model using a huge corpus of SMILES, a text representation system for molecules. We performed benchmarks on 10 datasets against existing fingerprints and graph-based methods and demonstrated the superiority of the proposed algorithms in small-data settings where pre-training facilitated good generalization. Moreover, we define a novel metric to concurrently measure model accuracy and data efficiency.
Self-Attention Based Molecule Representation for Predicting Drug-Target Interaction
Predicting drug-target interactions (DTI) is an essential part of the drug discovery process, which is an expensive process in terms of time and cost. Therefore, reducing DTI cost could lead to reduced healthcare costs for a patient. In addition, a precisely learned molecule representation in a DTI model could contribute to developing personalized medicine, which will help many patient cohorts. In this paper, we propose a new molecule representation based on the self-attention mechanism, and a new DTI model using our molecule representation. The experiments show that our DTI model outperforms the state of the art by up to 4.9% points in terms of area under the precision-recall curve. Moreover, a study using the DrugBank database proves that our model effectively lists all known drugs targeting a specific cancer biomarker in the top-30 candidate list.
MOROCO: The Moldavian and Romanian Dialectal Corpus
In this work, we introduce the MOldavian and ROmanian Dialectal COrpus (MOROCO), which is freely available for download at https://github.com/butnaruandrei/MOROCO. The corpus contains 33564 samples of text (with over 10 million tokens) collected from the news domain. The samples belong to one of the following six topics: culture, finance, politics, science, sports and tech. The data set is divided into 21719 samples for training, 5921 samples for validation and another 5924 samples for testing. For each sample, we provide corresponding dialectal and category labels. This allows us to perform empirical studies on several classification tasks such as (i) binary discrimination of Moldavian versus Romanian text samples, (ii) intra-dialect multi-class categorization by topic and (iii) cross-dialect multi-class categorization by topic. We perform experiments using a shallow approach based on string kernels, as well as a novel deep approach based on character-level convolutional neural networks containing Squeeze-and-Excitation blocks. We also present and analyze the most discriminative features of our best performing model, before and after named entity removal.
Molecular Sets (MOSES): A Benchmarking Platform for Molecular Generation Models
Generative models are becoming a tool of choice for exploring the molecular space. These models learn on a large training dataset and produce novel molecular structures with similar properties. Generated structures can be utilized for virtual screening or training semi-supervised predictive models in the downstream tasks. While there are plenty of generative models, it is unclear how to compare and rank them. In this work, we introduce a benchmarking platform called Molecular Sets (MOSES) to standardize training and comparison of molecular generative models. MOSES provides a training and testing datasets, and a set of metrics to evaluate the quality and diversity of generated structures. We have implemented and compared several molecular generation models and suggest to use our results as reference points for further advancements in generative chemistry research. The platform and source code are available at https://github.com/molecularsets/moses.


MoleculeNet: A Benchmark for Molecular Machine Learning
Molecular machine learning has been maturing rapidly over the last few years. Improved methods and the presence of larger datasets have enabled machine learning algorithms to make increasingly accurate predictions about molecular properties. However, algorithmic progress has been limited due to the lack of a standard benchmark to compare the efficacy of proposed methods; most new algorithms are benchmarked on different datasets making it challenging to gauge the quality of proposed methods. This work introduces MoleculeNet, a large scale benchmark for molecular machine learning. MoleculeNet curates multiple public datasets, establishes metrics for evaluation, and offers high quality open-source implementations of multiple previously proposed molecular featurization and learning algorithms (released as part of the DeepChem open source library). MoleculeNet benchmarks demonstrate that learnable representations are powerful tools for molecular machine learning and broadly offer the best performance. However, this result comes with caveats. Learnable representations still struggle to deal with complex tasks under data scarcity and highly imbalanced classification. For quantum mechanical and biophysical datasets, the use of physics-aware featurizations can be more important than choice of particular learning algorithm.
Stack More Layers Differently: High-Rank Training Through Low-Rank Updates
Despite the dominance and effectiveness of scaling, resulting in large networks with hundreds of billions of parameters, the necessity to train overparametrized models remains poorly understood, and alternative approaches do not necessarily make it cheaper to train high-performance models. In this paper, we explore low-rank training techniques as an alternative approach to training large neural networks. We introduce a novel method called ReLoRA, which utilizes low-rank updates to train high-rank networks. We apply ReLoRA to pre-training transformer language models with up to 350M parameters and demonstrate comparable performance to regular neural network training. Furthermore, we observe that the efficiency of ReLoRA increases with model size, making it a promising approach for training multi-billion-parameter networks efficiently. Our findings shed light on the potential of low-rank training techniques and their implications for scaling laws.

Learning Molecular Representation in a Cell
Predicting drug efficacy and safety in vivo requires information on biological responses (e.g., cell morphology and gene expression) to small molecule perturbations. However, current molecular representation learning methods do not provide a comprehensive view of cell states under these perturbations and struggle to remove noise, hindering model generalization. We introduce the Information Alignment (InfoAlign) approach to learn molecular representations through the information bottleneck method in cells. We integrate molecules and cellular response data as nodes into a context graph, connecting them with weighted edges based on chemical, biological, and computational criteria. For each molecule in a training batch, InfoAlign optimizes the encoder's latent representation with a minimality objective to discard redundant structural information. A sufficiency objective decodes the representation to align with different feature spaces from the molecule's neighborhood in the context graph. We demonstrate that the proposed sufficiency objective for alignment is tighter than existing encoder-based contrastive methods. Empirically, we validate representations from InfoAlign in two downstream tasks: molecular property prediction against up to 19 baseline methods across four datasets, plus zero-shot molecule-morphology matching.




3D molecule generation by denoising voxel grids
We propose a new score-based approach to generate 3D molecules represented as atomic densities on regular grids. First, we train a denoising neural network that learns to map from a smooth distribution of noisy molecules to the distribution of real molecules. Then, we follow the neural empirical Bayes framework [Saremi and Hyvarinen, 2019] and generate molecules in two steps: (i) sample noisy density grids from a smooth distribution via underdamped Langevin Markov chain Monte Carlo, and (ii) recover the ``clean'' molecule by denoising the noisy grid with a single step. Our method, VoxMol, generates molecules in a fundamentally different way than the current state of the art (i.e., diffusion models applied to atom point clouds). It differs in terms of the data representation, the noise model, the network architecture and the generative modeling algorithm. VoxMol achieves comparable results to state of the art on unconditional 3D molecule generation while being simpler to train and faster to generate molecules.
MolCA: Molecular Graph-Language Modeling with Cross-Modal Projector and Uni-Modal Adapter
Language Models (LMs) have demonstrated impressive molecule understanding ability on various 1D text-related tasks. However, they inherently lack 2D graph perception - a critical ability of human professionals in comprehending molecules' topological structures. To bridge this gap, we propose MolCA: Molecular Graph-Language Modeling with Cross-Modal Projector and Uni-Modal Adapter. MolCA enables an LM (e.g., Galactica) to understand both text- and graph-based molecular contents via the cross-modal projector. Specifically, the cross-modal projector is implemented as a Q-Former to connect a graph encoder's representation space and an LM's text space. Further, MolCA employs a uni-modal adapter (i.e., LoRA) for the LM's efficient adaptation to downstream tasks. Unlike previous studies that couple an LM with a graph encoder via cross-modal contrastive learning, MolCA retains the LM's ability of open-ended text generation and augments it with 2D graph information. To showcase its effectiveness, we extensively benchmark MolCA on tasks of molecule captioning, IUPAC name prediction, and molecule-text retrieval, on which MolCA significantly outperforms the baselines. Our codes and checkpoints can be found at https://github.com/acharkq/MolCA.

Extracting Molecular Properties from Natural Language with Multimodal Contrastive Learning
Deep learning in computational biochemistry has traditionally focused on molecular graphs neural representations; however, recent advances in language models highlight how much scientific knowledge is encoded in text. To bridge these two modalities, we investigate how molecular property information can be transferred from natural language to graph representations. We study property prediction performance gains after using contrastive learning to align neural graph representations with representations of textual descriptions of their characteristics. We implement neural relevance scoring strategies to improve text retrieval, introduce a novel chemically-valid molecular graph augmentation strategy inspired by organic reactions, and demonstrate improved performance on downstream MoleculeNet property classification tasks. We achieve a +4.26% AUROC gain versus models pre-trained on the graph modality alone, and a +1.54% gain compared to recently proposed molecular graph/text contrastively trained MoMu model (Su et al. 2022).

Towards More Effective and Economic Sparsely-Activated Model
The sparsely-activated models have achieved great success in natural language processing through large-scale parameters and relatively low computational cost, and gradually become a feasible technique for training and implementing extremely large models. Due to the limit of communication cost, activating multiple experts is hardly affordable during training and inference. Therefore, previous work usually activate just one expert at a time to alleviate additional communication cost. Such routing mechanism limits the upper bound of model performance. In this paper, we first investigate a phenomenon that increasing the number of activated experts can boost the model performance with higher sparse ratio. To increase the number of activated experts without an increase in computational cost, we propose SAM (Switch and Mixture) routing, an efficient hierarchical routing mechanism that activates multiple experts in a same device (GPU). Our methods shed light on the training of extremely large sparse models and experiments prove that our models can achieve significant performance gain with great efficiency improvement.

$\textit{L+M-24}$: Building a Dataset for Language + Molecules @ ACL 2024
Language-molecule models have emerged as an exciting direction for molecular discovery and understanding. However, training these models is challenging due to the scarcity of molecule-language pair datasets. At this point, datasets have been released which are 1) small and scraped from existing databases, 2) large but noisy and constructed by performing entity linking on the scientific literature, and 3) built by converting property prediction datasets to natural language using templates. In this document, we detail the L+M-24 dataset, which has been created for the Language + Molecules Workshop shared task at ACL 2024. In particular, L+M-24 is designed to focus on three key benefits of natural language in molecule design: compositionality, functionality, and abstraction.

Towards More Accurate Diffusion Model Acceleration with A Timestep Aligner
A diffusion model, which is formulated to produce an image using thousands of denoising steps, usually suffers from a slow inference speed. Existing acceleration algorithms simplify the sampling by skipping most steps yet exhibit considerable performance degradation. By viewing the generation of diffusion models as a discretized integrating process, we argue that the quality drop is partly caused by applying an inaccurate integral direction to a timestep interval. To rectify this issue, we propose a timestep aligner that helps find a more accurate integral direction for a particular interval at the minimum cost. Specifically, at each denoising step, we replace the original parameterization by conditioning the network on a new timestep, which is obtained by aligning the sampling distribution to the real distribution. Extensive experiments show that our plug-in design can be trained efficiently and boost the inference performance of various state-of-the-art acceleration methods, especially when there are few denoising steps. For example, when using 10 denoising steps on the popular LSUN Bedroom dataset, we improve the FID of DDIM from 9.65 to 6.07, simply by adopting our method for a more appropriate set of timesteps. Code will be made publicly available.
SELFormer: Molecular Representation Learning via SELFIES Language Models
Automated computational analysis of the vast chemical space is critical for numerous fields of research such as drug discovery and material science. Representation learning techniques have recently been employed with the primary objective of generating compact and informative numerical expressions of complex data. One approach to efficiently learn molecular representations is processing string-based notations of chemicals via natural language processing (NLP) algorithms. Majority of the methods proposed so far utilize SMILES notations for this purpose; however, SMILES is associated with numerous problems related to validity and robustness, which may prevent the model from effectively uncovering the knowledge hidden in the data. In this study, we propose SELFormer, a transformer architecture-based chemical language model that utilizes a 100% valid, compact and expressive notation, SELFIES, as input, in order to learn flexible and high-quality molecular representations. SELFormer is pre-trained on two million drug-like compounds and fine-tuned for diverse molecular property prediction tasks. Our performance evaluation has revealed that, SELFormer outperforms all competing methods, including graph learning-based approaches and SMILES-based chemical language models, on predicting aqueous solubility of molecules and adverse drug reactions. We also visualized molecular representations learned by SELFormer via dimensionality reduction, which indicated that even the pre-trained model can discriminate molecules with differing structural properties. We shared SELFormer as a programmatic tool, together with its datasets and pre-trained models. Overall, our research demonstrates the benefit of using the SELFIES notations in the context of chemical language modeling and opens up new possibilities for the design and discovery of novel drug candidates with desired features.

Astroformer: More Data Might not be all you need for Classification
Recent advancements in areas such as natural language processing and computer vision rely on intricate and massive models that have been trained using vast amounts of unlabelled or partly labeled data and training or deploying these state-of-the-art methods to resource constraint environments has been a challenge. Galaxy morphologies are crucial to understanding the processes by which galaxies form and evolve. Efficient methods to classify galaxy morphologies are required to extract physical information from modern-day astronomy surveys. In this paper, we introduce Astroformer, a method to learn from less amount of data. We propose using a hybrid transformer-convolutional architecture drawing much inspiration from the success of CoAtNet and MaxViT. Concretely, we use the transformer-convolutional hybrid with a new stack design for the network, a different way of creating a relative self-attention layer, and pair it with a careful selection of data augmentation and regularization techniques. Our approach sets a new state-of-the-art on predicting galaxy morphologies from images on the Galaxy10 DECals dataset, a science objective, which consists of 17736 labeled images achieving 94.86% top-1 accuracy, beating the current state-of-the-art for this task by 4.62%. Furthermore, this approach also sets a new state-of-the-art on CIFAR-100 and Tiny ImageNet. We also find that models and training methods used for larger datasets would often not work very well in the low-data regime.

ClassDiffusion: More Aligned Personalization Tuning with Explicit Class Guidance
Recent text-to-image customization works have been proven successful in generating images of given concepts by fine-tuning the diffusion models on a few examples. However, these methods tend to overfit the concepts, resulting in failure to create the concept under multiple conditions (e.g. headphone is missing when generating a <sks> dog wearing a headphone'). Interestingly, we notice that the base model before fine-tuning exhibits the capability to compose the base concept with other elements (e.g. a dog wearing a headphone) implying that the compositional ability only disappears after personalization tuning. Inspired by this observation, we present ClassDiffusion, a simple technique that leverages a semantic preservation loss to explicitly regulate the concept space when learning the new concept. Despite its simplicity, this helps avoid semantic drift when fine-tuning on the target concepts. Extensive qualitative and quantitative experiments demonstrate that the use of semantic preservation loss effectively improves the compositional abilities of the fine-tune models. In response to the ineffective evaluation of CLIP-T metrics, we introduce BLIP2-T metric, a more equitable and effective evaluation metric for this particular domain. We also provide in-depth empirical study and theoretical analysis to better understand the role of the proposed loss. Lastly, we also extend our ClassDiffusion to personalized video generation, demonstrating its flexibility.

Say More with Less: Understanding Prompt Learning Behaviors through Gist Compression
Large language models (LLMs) require lengthy prompts as the input context to produce output aligned with user intentions, a process that incurs extra costs during inference. In this paper, we propose the Gist COnditioned deCOding (Gist-COCO) model, introducing a novel method for compressing prompts which also can assist the prompt interpretation and engineering. Gist-COCO employs an encoder-decoder based language model and then incorporates an additional encoder as a plugin module to compress prompts with inputs using gist tokens. It finetunes the compression plugin module and uses the representations of gist tokens to emulate the raw prompts in the vanilla language model. By verbalizing the representations of gist tokens into gist prompts, the compression ability of Gist-COCO can be generalized to different LLMs with high compression rates. Our experiments demonstrate that Gist-COCO outperforms previous prompt compression models in both passage and instruction compression tasks. Further analysis on gist verbalization results suggests that our gist prompts serve different functions in aiding language models. They may directly provide potential answers, generate the chain-of-thought, or simply repeat the inputs. All data and codes are available at https://github.com/OpenMatch/Gist-COCO .

Get More with LESS: Synthesizing Recurrence with KV Cache Compression for Efficient LLM Inference
Many computational factors limit broader deployment of large language models. In this paper, we focus on a memory bottleneck imposed by the key-value (KV) cache, a computational shortcut that requires storing previous KV pairs during decoding. While existing KV cache methods approach this problem by pruning or evicting large swaths of relatively less important KV pairs to dramatically reduce the memory footprint of the cache, they can have limited success in tasks that require recollecting a majority of previous tokens. To alleviate this issue, we propose LESS, a simple integration of a (nearly free) constant sized cache with eviction-based cache methods, such that all tokens can be queried at later decoding steps. Its ability to retain information throughout time shows merit on a variety of tasks where we demonstrate LESS can help reduce the performance gap from caching everything, sometimes even matching it, all while being efficient.


Scientific Language Modeling: A Quantitative Review of Large Language Models in Molecular Science
Efficient molecular modeling and design are crucial for the discovery and exploration of novel molecules, and the incorporation of deep learning methods has revolutionized this field. In particular, large language models (LLMs) offer a fresh approach to tackle scientific problems from a natural language processing (NLP) perspective, introducing a research paradigm called scientific language modeling (SLM). However, two key issues remain: how to quantify the match between model and data modalities and how to identify the knowledge-learning preferences of models. To address these challenges, we propose a multi-modal benchmark, named ChEBI-20-MM, and perform 1263 experiments to assess the model's compatibility with data modalities and knowledge acquisition. Through the modal transition probability matrix, we provide insights into the most suitable modalities for tasks. Furthermore, we introduce a statistically interpretable approach to discover context-specific knowledge mapping by localized feature filtering. Our pioneering analysis offers an exploration of the learning mechanism and paves the way for advancing SLM in molecular science.
See More Details: Efficient Image Super-Resolution by Experts Mining
Reconstructing high-resolution (HR) images from low-resolution (LR) inputs poses a significant challenge in image super-resolution (SR). While recent approaches have demonstrated the efficacy of intricate operations customized for various objectives, the straightforward stacking of these disparate operations can result in a substantial computational burden, hampering their practical utility. In response, we introduce SeemoRe, an efficient SR model employing expert mining. Our approach strategically incorporates experts at different levels, adopting a collaborative methodology. At the macro scale, our experts address rank-wise and spatial-wise informative features, providing a holistic understanding. Subsequently, the model delves into the subtleties of rank choice by leveraging a mixture of low-rank experts. By tapping into experts specialized in distinct key factors crucial for accurate SR, our model excels in uncovering intricate intra-feature details. This collaborative approach is reminiscent of the concept of "see more", allowing our model to achieve an optimal performance with minimal computational costs in efficient settings. The source will be publicly made available at https://github.com/eduardzamfir/seemoredetails
Towards More Unified In-context Visual Understanding
The rapid advancement of large language models (LLMs) has accelerated the emergence of in-context learning (ICL) as a cutting-edge approach in the natural language processing domain. Recently, ICL has been employed in visual understanding tasks, such as semantic segmentation and image captioning, yielding promising results. However, existing visual ICL framework can not enable producing content across multiple modalities, which limits their potential usage scenarios. To address this issue, we present a new ICL framework for visual understanding with multi-modal output enabled. First, we quantize and embed both text and visual prompt into a unified representational space, structured as interleaved in-context sequences. Then a decoder-only sparse transformer architecture is employed to perform generative modeling on them, facilitating in-context learning. Thanks to this design, the model is capable of handling in-context vision understanding tasks with multimodal output in a unified pipeline. Experimental results demonstrate that our model achieves competitive performance compared with specialized models and previous ICL baselines. Overall, our research takes a further step toward unified multimodal in-context learning.
Generating Molecular Conformer Fields
In this paper we tackle the problem of generating conformers of a molecule in 3D space given its molecular graph. We parameterize these conformers as continuous functions that map elements from the molecular graph to points in 3D space. We then formulate the problem of learning to generate conformers as learning a distribution over these functions using a diffusion generative model, called Molecular Conformer Fields (MCF). Our approach is simple and scalable, and achieves state-of-the-art performance on challenging molecular conformer generation benchmarks while making no assumptions about the explicit structure of molecules (e.g. modeling torsional angles). MCF represents an advance in extending diffusion models to handle complex scientific problems in a conceptually simple, scalable and effective manner.

One More Step: A Versatile Plug-and-Play Module for Rectifying Diffusion Schedule Flaws and Enhancing Low-Frequency Controls
It is well known that many open-released foundational diffusion models have difficulty in generating images that substantially depart from average brightness, despite such images being present in the training data. This is due to an inconsistency: while denoising starts from pure Gaussian noise during inference, the training noise schedule retains residual data even in the final timestep distribution, due to difficulties in numerical conditioning in mainstream formulation, leading to unintended bias during inference. To mitigate this issue, certain epsilon-prediction models are combined with an ad-hoc offset-noise methodology. In parallel, some contemporary models have adopted zero-terminal SNR noise schedules together with v-prediction, which necessitate major alterations to pre-trained models. However, such changes risk destabilizing a large multitude of community-driven applications anchored on these pre-trained models. In light of this, our investigation revisits the fundamental causes, leading to our proposal of an innovative and principled remedy, called One More Step (OMS). By integrating a compact network and incorporating an additional simple yet effective step during inference, OMS elevates image fidelity and harmonizes the dichotomy between training and inference, while preserving original model parameters. Once trained, various pre-trained diffusion models with the same latent domain can share the same OMS module.

Sliced Denoising: A Physics-Informed Molecular Pre-Training Method
While molecular pre-training has shown great potential in enhancing drug discovery, the lack of a solid physical interpretation in current methods raises concerns about whether the learned representation truly captures the underlying explanatory factors in observed data, ultimately resulting in limited generalization and robustness. Although denoising methods offer a physical interpretation, their accuracy is often compromised by ad-hoc noise design, leading to inaccurate learned force fields. To address this limitation, this paper proposes a new method for molecular pre-training, called sliced denoising (SliDe), which is based on the classical mechanical intramolecular potential theory. SliDe utilizes a novel noise strategy that perturbs bond lengths, angles, and torsion angles to achieve better sampling over conformations. Additionally, it introduces a random slicing approach that circumvents the computationally expensive calculation of the Jacobian matrix, which is otherwise essential for estimating the force field. By aligning with physical principles, SliDe shows a 42\% improvement in the accuracy of estimated force fields compared to current state-of-the-art denoising methods, and thus outperforms traditional baselines on various molecular property prediction tasks.
From Molecules to Materials: Pre-training Large Generalizable Models for Atomic Property Prediction
Foundation models have been transformational in machine learning fields such as natural language processing and computer vision. Similar success in atomic property prediction has been limited due to the challenges of training effective models across multiple chemical domains. To address this, we introduce Joint Multi-domain Pre-training (JMP), a supervised pre-training strategy that simultaneously trains on multiple datasets from different chemical domains, treating each dataset as a unique pre-training task within a multi-task framework. Our combined training dataset consists of sim120M systems from OC20, OC22, ANI-1x, and Transition-1x. We evaluate performance and generalization by fine-tuning over a diverse set of downstream tasks and datasets including: QM9, rMD17, MatBench, QMOF, SPICE, and MD22. JMP demonstrates an average improvement of 59% over training from scratch, and matches or sets state-of-the-art on 34 out of 40 tasks. Our work highlights the potential of pre-training strategies that utilize diverse data to advance property prediction across chemical domains, especially for low-data tasks.


Gotta be SAFE: A New Framework for Molecular Design
Traditional molecular string representations, such as SMILES, often pose challenges for AI-driven molecular design due to their non-sequential depiction of molecular substructures. To address this issue, we introduce Sequential Attachment-based Fragment Embedding (SAFE), a novel line notation for chemical structures. SAFE reimagines SMILES strings as an unordered sequence of interconnected fragment blocks while maintaining full compatibility with existing SMILES parsers. It streamlines complex generative tasks, including scaffold decoration, fragment linking, polymer generation, and scaffold hopping, while facilitating autoregressive generation for fragment-constrained design, thereby eliminating the need for intricate decoding or graph-based models. We demonstrate the effectiveness of SAFE by training an 87-million-parameter GPT2-like model on a dataset containing 1.1 billion SAFE representations. Through extensive experimentation, we show that our SAFE-GPT model exhibits versatile and robust optimization performance. SAFE opens up new avenues for the rapid exploration of chemical space under various constraints, promising breakthroughs in AI-driven molecular design.


Towards Foundational Models for Molecular Learning on Large-Scale Multi-Task Datasets
Recently, pre-trained foundation models have enabled significant advancements in multiple fields. In molecular machine learning, however, where datasets are often hand-curated, and hence typically small, the lack of datasets with labeled features, and codebases to manage those datasets, has hindered the development of foundation models. In this work, we present seven novel datasets categorized by size into three distinct categories: ToyMix, LargeMix and UltraLarge. These datasets push the boundaries in both the scale and the diversity of supervised labels for molecular learning. They cover nearly 100 million molecules and over 3000 sparsely defined tasks, totaling more than 13 billion individual labels of both quantum and biological nature. In comparison, our datasets contain 300 times more data points than the widely used OGB-LSC PCQM4Mv2 dataset, and 13 times more than the quantum-only QM1B dataset. In addition, to support the development of foundational models based on our proposed datasets, we present the Graphium graph machine learning library which simplifies the process of building and training molecular machine learning models for multi-task and multi-level molecular datasets. Finally, we present a range of baseline results as a starting point of multi-task and multi-level training on these datasets. Empirically, we observe that performance on low-resource biological datasets show improvement by also training on large amounts of quantum data. This indicates that there may be potential in multi-task and multi-level training of a foundation model and fine-tuning it to resource-constrained downstream tasks.

Beam Enumeration: Probabilistic Explainability For Sample Efficient Self-conditioned Molecular Design
Generative molecular design has moved from proof-of-concept to real-world applicability, as marked by the surge in very recent papers reporting experimental validation. Key challenges in explainability and sample efficiency present opportunities to enhance generative design to directly optimize expensive high-fidelity oracles and provide actionable insights to domain experts. Here, we propose Beam Enumeration to exhaustively enumerate the most probable sub-sequences from language-based molecular generative models and show that molecular substructures can be extracted. When coupled with reinforcement learning, extracted substructures become meaningful, providing a source of explainability and improving sample efficiency through self-conditioned generation. Beam Enumeration is generally applicable to any language-based molecular generative model and notably further improves the performance of the recently reported Augmented Memory algorithm, which achieved the new state-of-the-art on the Practical Molecular Optimization benchmark for sample efficiency. The combined algorithm generates more high reward molecules and faster, given a fixed oracle budget. Beam Enumeration shows that improvements to explainability and sample efficiency for molecular design can be made synergistic.
See More and Know More: Zero-shot Point Cloud Segmentation via Multi-modal Visual Data
Zero-shot point cloud segmentation aims to make deep models capable of recognizing novel objects in point cloud that are unseen in the training phase. Recent trends favor the pipeline which transfers knowledge from seen classes with labels to unseen classes without labels. They typically align visual features with semantic features obtained from word embedding by the supervision of seen classes' annotations. However, point cloud contains limited information to fully match with semantic features. In fact, the rich appearance information of images is a natural complement to the textureless point cloud, which is not well explored in previous literature. Motivated by this, we propose a novel multi-modal zero-shot learning method to better utilize the complementary information of point clouds and images for more accurate visual-semantic alignment. Extensive experiments are performed in two popular benchmarks, i.e., SemanticKITTI and nuScenes, and our method outperforms current SOTA methods with 52% and 49% improvement on average for unseen class mIoU, respectively.
Multimodal Molecular Pretraining via Modality Blending
Self-supervised learning has recently gained growing interest in molecular modeling for scientific tasks such as AI-assisted drug discovery. Current studies consider leveraging both 2D and 3D molecular structures for representation learning. However, relying on straightforward alignment strategies that treat each modality separately, these methods fail to exploit the intrinsic correlation between 2D and 3D representations that reflect the underlying structural characteristics of molecules, and only perform coarse-grained molecule-level alignment. To derive fine-grained alignment and promote structural molecule understanding, we introduce an atomic-relation level "blend-then-predict" self-supervised learning approach, MoleBLEND, which first blends atom relations represented by different modalities into one unified relation matrix for joint encoding, then recovers modality-specific information for 2D and 3D structures individually. By treating atom relationships as anchors, MoleBLEND organically aligns and integrates visually dissimilar 2D and 3D modalities of the same molecule at fine-grained atomic level, painting a more comprehensive depiction of each molecule. Extensive experiments show that MoleBLEND achieves state-of-the-art performance across major 2D/3D molecular benchmarks. We further provide theoretical insights from the perspective of mutual-information maximization, demonstrating that our method unifies contrastive, generative (cross-modality prediction) and mask-then-predict (single-modality prediction) objectives into one single cohesive framework.
Towards More Realistic Membership Inference Attacks on Large Diffusion Models
Generative diffusion models, including Stable Diffusion and Midjourney, can generate visually appealing, diverse, and high-resolution images for various applications. These models are trained on billions of internet-sourced images, raising significant concerns about the potential unauthorized use of copyright-protected images. In this paper, we examine whether it is possible to determine if a specific image was used in the training set, a problem known in the cybersecurity community and referred to as a membership inference attack. Our focus is on Stable Diffusion, and we address the challenge of designing a fair evaluation framework to answer this membership question. We propose a methodology to establish a fair evaluation setup and apply it to Stable Diffusion, enabling potential extensions to other generative models. Utilizing this evaluation setup, we execute membership attacks (both known and newly introduced). Our research reveals that previously proposed evaluation setups do not provide a full understanding of the effectiveness of membership inference attacks. We conclude that the membership inference attack remains a significant challenge for large diffusion models (often deployed as black-box systems), indicating that related privacy and copyright issues will persist in the foreseeable future.

Unifying Molecular and Textual Representations via Multi-task Language Modelling
The recent advances in neural language models have also been successfully applied to the field of chemistry, offering generative solutions for classical problems in molecular design and synthesis planning. These new methods have the potential to optimize laboratory operations and fuel a new era of data-driven automation in scientific discovery. However, specialized models are still typically required for each task, leading to the need for problem-specific fine-tuning and neglecting task interrelations. The main obstacle in this field is the lack of a unified representation between natural language and chemical representations, complicating and limiting human-machine interaction. Here, we propose a multi-domain, multi-task language model to solve a wide range of tasks in both the chemical and natural language domains. By leveraging multi-task learning, our model can handle chemical and natural language concurrently, without requiring expensive pre-training on single domains or task-specific models. Interestingly, sharing weights across domains remarkably improves our model when benchmarked against state-of-the-art baselines on single-domain and cross-domain tasks. In particular, sharing information across domains and tasks gives rise to large improvements in cross-domain tasks, the magnitude of which increase with scale, as measured by more than a dozen of relevant metrics. Our work suggests that such models can robustly and efficiently accelerate discovery in physical sciences by superseding problem-specific fine-tuning and enhancing human-model interactions.


Mask More and Mask Later: Efficient Pre-training of Masked Language Models by Disentangling the [MASK] Token
The pre-training of masked language models (MLMs) consumes massive computation to achieve good results on downstream NLP tasks, resulting in a large carbon footprint. In the vanilla MLM, the virtual tokens, [MASK]s, act as placeholders and gather the contextualized information from unmasked tokens to restore the corrupted information. It raises the question of whether we can append [MASK]s at a later layer, to reduce the sequence length for earlier layers and make the pre-training more efficient. We show: (1) [MASK]s can indeed be appended at a later layer, being disentangled from the word embedding; (2) The gathering of contextualized information from unmasked tokens can be conducted with a few layers. By further increasing the masking rate from 15% to 50%, we can pre-train RoBERTa-base and RoBERTa-large from scratch with only 78% and 68% of the original computational budget without any degradation on the GLUE benchmark. When pre-training with the original budget, our method outperforms RoBERTa for 6 out of 8 GLUE tasks, on average by 0.4%.

The More Secure, The Less Equally Usable: Gender and Ethnicity (Un)fairness of Deep Face Recognition along Security Thresholds
Face biometrics are playing a key role in making modern smart city applications more secure and usable. Commonly, the recognition threshold of a face recognition system is adjusted based on the degree of security for the considered use case. The likelihood of a match can be for instance decreased by setting a high threshold in case of a payment transaction verification. Prior work in face recognition has unfortunately showed that error rates are usually higher for certain demographic groups. These disparities have hence brought into question the fairness of systems empowered with face biometrics. In this paper, we investigate the extent to which disparities among demographic groups change under different security levels. Our analysis includes ten face recognition models, three security thresholds, and six demographic groups based on gender and ethnicity. Experiments show that the higher the security of the system is, the higher the disparities in usability among demographic groups are. Compelling unfairness issues hence exist and urge countermeasures in real-world high-stakes environments requiring severe security levels.
FusionRetro: Molecule Representation Fusion via In-Context Learning for Retrosynthetic Planning
Retrosynthetic planning aims to devise a complete multi-step synthetic route from starting materials to a target molecule. Current strategies use a decoupled approach of single-step retrosynthesis models and search algorithms, taking only the product as the input to predict the reactants for each planning step and ignoring valuable context information along the synthetic route. In this work, we propose a novel framework that utilizes context information for improved retrosynthetic planning. We view synthetic routes as reaction graphs and propose to incorporate context through three principled steps: encode molecules into embeddings, aggregate information over routes, and readout to predict reactants. Our approach is the first attempt to utilize in-context learning for retrosynthesis prediction in retrosynthetic planning. The entire framework can be efficiently optimized in an end-to-end fashion and produce more practical and accurate predictions. Comprehensive experiments demonstrate that by fusing in the context information over routes, our model significantly improves the performance of retrosynthetic planning over baselines that are not context-aware, especially for long synthetic routes. Code is available at https://github.com/SongtaoLiu0823/FusionRetro.


No More Strided Convolutions or Pooling: A New CNN Building Block for Low-Resolution Images and Small Objects
Convolutional neural networks (CNNs) have made resounding success in many computer vision tasks such as image classification and object detection. However, their performance degrades rapidly on tougher tasks where images are of low resolution or objects are small. In this paper, we point out that this roots in a defective yet common design in existing CNN architectures, namely the use of strided convolution and/or pooling layers, which results in a loss of fine-grained information and learning of less effective feature representations. To this end, we propose a new CNN building block called SPD-Conv in place of each strided convolution layer and each pooling layer (thus eliminates them altogether). SPD-Conv is comprised of a space-to-depth (SPD) layer followed by a non-strided convolution (Conv) layer, and can be applied in most if not all CNN architectures. We explain this new design under two most representative computer vision tasks: object detection and image classification. We then create new CNN architectures by applying SPD-Conv to YOLOv5 and ResNet, and empirically show that our approach significantly outperforms state-of-the-art deep learning models, especially on tougher tasks with low-resolution images and small objects. We have open-sourced our code at https://github.com/LabSAINT/SPD-Conv.
Is More Data All You Need? A Causal Exploration
Curating a large scale medical imaging dataset for machine learning applications is both time consuming and expensive. Balancing the workload between model development, data collection and annotations is difficult for machine learning practitioners, especially under time constraints. Causal analysis is often used in medicine and economics to gain insights about the effects of actions and policies. In this paper we explore the effect of dataset interventions on the output of image classification models. Through a causal approach we investigate the effects of the quantity and type of data we need to incorporate in a dataset to achieve better performance for specific subtasks. The main goal of this paper is to highlight the potential of causal analysis as a tool for resource optimization for developing medical imaging ML applications. We explore this concept with a synthetic dataset and an exemplary use-case for Diabetic Retinopathy image analysis.
Relative Molecule Self-Attention Transformer
Self-supervised learning holds promise to revolutionize molecule property prediction - a central task to drug discovery and many more industries - by enabling data efficient learning from scarce experimental data. Despite significant progress, non-pretrained methods can be still competitive in certain settings. We reason that architecture might be a key bottleneck. In particular, enriching the backbone architecture with domain-specific inductive biases has been key for the success of self-supervised learning in other domains. In this spirit, we methodologically explore the design space of the self-attention mechanism tailored to molecular data. We identify a novel variant of self-attention adapted to processing molecules, inspired by the relative self-attention layer, which involves fusing embedded graph and distance relationships between atoms. Our main contribution is Relative Molecule Attention Transformer (R-MAT): a novel Transformer-based model based on the developed self-attention layer that achieves state-of-the-art or very competitive results across a~wide range of molecule property prediction tasks.
Be More Active! Understanding the Differences between Mean and Sampled Representations of Variational Autoencoders
The ability of Variational Autoencoders to learn disentangled representations has made them appealing for practical applications. However, their mean representations, which are generally used for downstream tasks, have recently been shown to be more correlated than their sampled counterpart, on which disentanglement is usually measured. In this paper, we refine this observation through the lens of selective posterior collapse, which states that only a subset of the learned representations, the active variables, is encoding useful information while the rest (the passive variables) is discarded. We first extend the existing definition to multiple data examples and show that active variables are equally disentangled in mean and sampled representations. Based on this extension and the pre-trained models from disentanglement lib, we then isolate the passive variables and show that they are responsible for the discrepancies between mean and sampled representations. Specifically, passive variables exhibit high correlation scores with other variables in mean representations while being fully uncorrelated in sampled ones. We thus conclude that despite what their higher correlation might suggest, mean representations are still good candidates for downstream tasks applications. However, it may be beneficial to remove their passive variables, especially when used with models sensitive to correlated features.
GhostNet: More Features from Cheap Operations
Deploying convolutional neural networks (CNNs) on embedded devices is difficult due to the limited memory and computation resources. The redundancy in feature maps is an important characteristic of those successful CNNs, but has rarely been investigated in neural architecture design. This paper proposes a novel Ghost module to generate more feature maps from cheap operations. Based on a set of intrinsic feature maps, we apply a series of linear transformations with cheap cost to generate many ghost feature maps that could fully reveal information underlying intrinsic features. The proposed Ghost module can be taken as a plug-and-play component to upgrade existing convolutional neural networks. Ghost bottlenecks are designed to stack Ghost modules, and then the lightweight GhostNet can be easily established. Experiments conducted on benchmarks demonstrate that the proposed Ghost module is an impressive alternative of convolution layers in baseline models, and our GhostNet can achieve higher recognition performance (e.g. 75.7% top-1 accuracy) than MobileNetV3 with similar computational cost on the ImageNet ILSVRC-2012 classification dataset. Code is available at https://github.com/huawei-noah/ghostnet

Know More about Each Other: Evolving Dialogue Strategy via Compound Assessment
In this paper, a novel Generation-Evaluation framework is developed for multi-turn conversations with the objective of letting both participants know more about each other. For the sake of rational knowledge utilization and coherent conversation flow, a dialogue strategy which controls knowledge selection is instantiated and continuously adapted via reinforcement learning. Under the deployed strategy, knowledge grounded conversations are conducted with two dialogue agents. The generated dialogues are comprehensively evaluated on aspects like informativeness and coherence, which are aligned with our objective and human instinct. These assessments are integrated as a compound reward to guide the evolution of dialogue strategy via policy gradient. Comprehensive experiments have been carried out on the publicly available dataset, demonstrating that the proposed method outperforms the other state-of-the-art approaches significantly.
Jet-ISM Interaction in the Radio Galaxy 3C293: Jet-driven Shocks Heat ISM to Power X-ray and Molecular H2 emission
We present a 70ks Chandra observation of the radio galaxy 3C293. This galaxy belongs to the class of molecular hydrogen emission galaxies (MOHEGs) that have very luminous emission from warm molecular hydrogen. In radio galaxies, the molecular gas appears to be heated by jet-driven shocks, but exactly how this mechanism works is still poorly understood. With Chandra, we observe X-ray emission from the jets within the host galaxy and along the 100 kpc radio jets. We model the X-ray spectra of the nucleus, the inner jets, and the X-ray features along the extended radio jets. Both the nucleus and the inner jets show evidence of 10^7 K shock-heated gas. The kinetic power of the jets is more than sufficient to heat the X-ray emitting gas within the host galaxy. The thermal X-ray and warm H2 luminosities of 3C293 are similar, indicating similar masses of X-ray hot gas and warm molecular gas. This is consistent with a picture where both derive from a multiphase, shocked interstellar medium (ISM). We find that radio-loud MOHEGs that are not brightest cluster galaxies (BCGs), like 3C293, typically have LH2/LX~1 and MH2/MX~1, whereas MOHEGs that are BCGs have LH2/LX~0.01 and MH2/MX~0.01. The more massive, virialized, hot atmosphere in BCGs overwhelms any direct X-ray emission from current jet-ISM interaction. On the other hand, LH2/LX~1 in the Spiderweb BCG at z=2, which resides in an unvirialized protocluster and hosts a powerful radio source. Over time, jet-ISM interaction may contribute to the establishment of a hot atmosphere in BCGs and other massive elliptical galaxies.
Reward-Consistent Dynamics Models are Strongly Generalizable for Offline Reinforcement Learning
Learning a precise dynamics model can be crucial for offline reinforcement learning, which, unfortunately, has been found to be quite challenging. Dynamics models that are learned by fitting historical transitions often struggle to generalize to unseen transitions. In this study, we identify a hidden but pivotal factor termed dynamics reward that remains consistent across transitions, offering a pathway to better generalization. Therefore, we propose the idea of reward-consistent dynamics models: any trajectory generated by the dynamics model should maximize the dynamics reward derived from the data. We implement this idea as the MOREC (Model-based Offline reinforcement learning with Reward Consistency) method, which can be seamlessly integrated into previous offline model-based reinforcement learning (MBRL) methods. MOREC learns a generalizable dynamics reward function from offline data, which is subsequently employed as a transition filter in any offline MBRL method: when generating transitions, the dynamics model generates a batch of transitions and selects the one with the highest dynamics reward value. On a synthetic task, we visualize that MOREC has a strong generalization ability and can surprisingly recover some distant unseen transitions. On 21 offline tasks in D4RL and NeoRL benchmarks, MOREC improves the previous state-of-the-art performance by a significant margin, i.e., 4.6% on D4RL tasks and 25.9% on NeoRL tasks. Notably, MOREC is the first method that can achieve above 95% online RL performance in 6 out of 12 D4RL tasks and 3 out of 9 NeoRL tasks.